AggV3 code book - functional annotation queries¶
Functional annotation VCFs include annotation from VEP115 with all human-relevant plugins. You will find the functional variant VCFs, organised by shard, at:
s3://019847484957-germline-aggregate-v3-supporting-data-landing/functional-annotation_2025-12-24
Querying the Functional annotation VCFs requires the following steps:
- Identify the correct subshard for your analysis.
- Query the functional annotation.
1. Identify the correct subshard for your analysis¶
There are two ways to identify the relevant subshards for your analysis:
- You can use the shard lookup tool to pull out the shards by inputting a locus. The BED file for the functional annotation shards is at:
s3://512426816668-gel-data-resources/dragen3.7.8/AggV3_resources/manifests/functional_annotation/2025-12-24/functional_annotation_shards.bed - Query the shard BED files with bedtools.
Bear in mind that you may need to look up the gene on Ensembl to get the location first.
2. Query the functional annotation¶
Now you can intersect query the subshard VCF in an interactive session or as a bash script. All the following queries use bcftools and split-vep to query and parse the functional annotation from VEP.
You will need to load bcftools in your terminal in your interactive session. You can do this easily using conda:
conda install bcftools
Filepaths
The following queries assume you have mounted only the relevant subshard VCF and index to your interactive session. If you have mounted the entire folder, you will need to modify the filepaths in the queries.
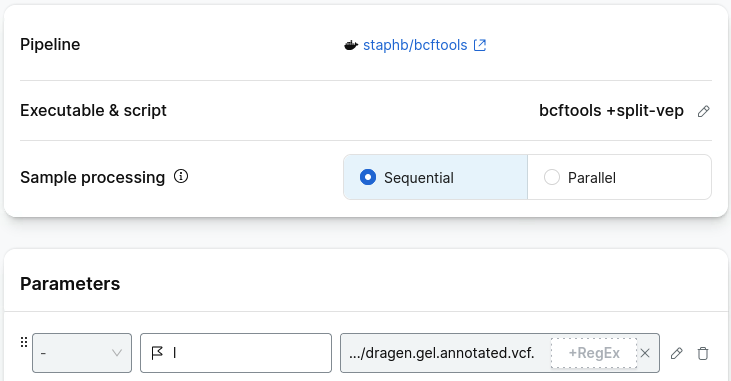
You will need to load bcftools as a container.
- Go to Batch analysis and select Run Pipeline.
- Search for bcftools and select a bcftools container
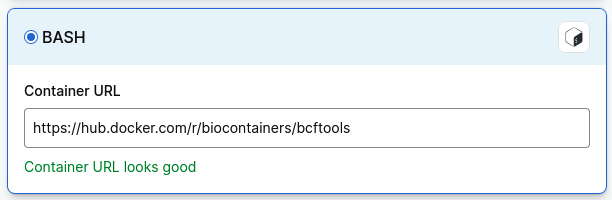
If you cannot find a bcftools container, select Import, then Bash and paste in the path to a bcftools container:
See what fields you can query¶
Split-vep allows you to view a list of all the fields created by VEP as part of our functional annotation analysis.
bcftools +split-vep -l mounted-data-readonly/dragen.gel.annotated.vcf.gz
Output:
The output is a list of fields added to the INFO column by VEP, which you can use to query the VCF.
...
1 Consequence
2 IMPACT
3 SYMBOL
4 Gene
5 Feature_type
6 Feature
7 BIOTYPE
8 EXON
9 INTRON
10 HGVSc
...
Extract variants above threshold for aggV3 derived allele frequencies¶
Question: I want to find common variants in genetically inferred European samples
Script: Use bcftools query with the -i flag to specify the allele frequency cut-off, and the -f flag to determine the output attributes and format.
Output: The first five lines are printed to the standard output.
...
chr1 10108 C T 0.073
chr1 10109 A T 0.083
chr1 10147 C A 0.063
chr1 10150 C T 0.067
chr1 10177 A C 0.132
...
Extract variants for a gene of interest¶
Question: I want to extract all variants in the gene IKZF1 and view some basic annotation.
Script: This example will output variants annotated against all transcripts for the gene of interest - IKZF1 - using the -i flag. There will be one annotation per line (for each transcript - using the -d flag). The -f option formats the output with the attributes included. The > character writes the output to a tab-delimited file.
Select executable script and add the follow as a shell script:
#!/bin/bash
vcf=$1
gene=$2
output=$3
bcftools +split-vep $vcf \
-i "SYMBOL=\"$gene\"" -d \
-f '%CHROM\t%POS\t%REF\t%ALT\t%SYMBOL\t%Feature\t%Consequence\t%Existing_variation\n' > $output
Add the parameters:
- the relevant shard VCF file
- your gene name of interest
- the output file name
For example:

Choose your project and run analysis.
Output: The output is a tab-delimited file in long-format - where each annotation is on a separate line across all variants. The columns are in the same order as stated in the -f command above.
...
chr7 50319076 G A IKZF1 ENST00000641948 non_coding_transcript_exon_variant rs374267123
chr7 50319076 G A IKZF1 ENST00000642219 synonymous_variant rs374267123
chr7 50319076 G A IKZF1 ENST00000645066 synonymous_variant rs374267123
chr7 50319076 G A IKZF1 ENST00000646110 synonymous_variant rs374267123
chr7 50319076 G C IKZF1 ENST00000331340 missense_variant rs374267123
chr7 50319076 G C IKZF1 ENST00000343574 missense_variant rs374267123
...
Extract variants for a gene of interest with a damaging prediction¶
Question: I want to view the variants that that are missense or worse and rare in gnomAD in the gene IKZF1.
Script: Use bcftools split-vep. This example will output variants annotated against all transcripts for the gene of interest - IKZF1 - that are missense or worse (-s and -S flag) and rare in Europeans (use the -c flag for numeric conversion). There will be one annotation per line (for each transcript - using the -d flag). The -f option formats the output with the attributes included. The > character writes the output to a tab-delimited file.
You will need to mount the VEP severity scale to your interactive session, you can find this at: s3 bucket
Select executable script and add the follow as a shell script:
#!/bin/bash
vcf=$1
gene=$2
sev=$3
output=$4
bcftools +split-vep $vcf \
-i "SYMBOL=\"$gene\" & EUR_AF<0.05" -d \
-f '%CHROM\t%POS\t%REF\t%ALT\t%SYMBOL\t%Feature\t%Consequence\t%Existing_variation\t%EUR_AF\n' \
-c SYMBOL,Feature,Consequence,EUR_AF:Float,Existing_variation \
-s worst:missense_variant+ -S $sev > $output
Add the parameters:
- the relevant shard VCF file
- your gene name of interest
- the severity scale file
- the output file name
For example:

Choose your project and run analysis.
Output: The output is a tab-delimited file in long-format - where each annotation is on a separate line across all variants. The columns are in the same order as stated in the -f command above.
...
chr7 50368156 C T IKZF1 ENST00000413698 missense_variant rs558055360 0
chr7 50368201 A G IKZF1 ENST00000413698 missense_variant rs117111762 0.002
chr7 50368251 A G IKZF1 ENST00000413698 missense_variant rs573829014 0
chr7 50368281 G A IKZF1 ENST00000413698 missense_variant rs562525663 0
chr7 50368296 G A IKZF1 ENST00000413698 missense_variant rs544990441 0
chr7 50376578 G A IKZF1 ENST00000331340 missense_variant rs144637662&COSM6972304 0.001
chr7 50376689 G A IKZF1 ENST00000331340 missense_variant rs549930725 0.001
chr7 50400076 G A IKZF1 ENST00000331340 missense_variant rs148169768 0
chr7 50400355 G C IKZF1 ENST00000331340 missense_variant rs529231990 0
...
Additional annotation queries¶
Below are some additional queries using split-vep that extract useful annotation.