AggV2 gVCF Aggregation¶
Single sample gVCFs were aggregated using gvcfgenotyper (Illumina, version: 2019.02.26), with post processing with bcftools and vt to create the multi-sample VCF of 78,195 samples. The aggregation workflow itself was written in the WDL language and executed using Cromwell.
The samples included in the aggregate can be found in the LabKey table aggregate_gvcf_sample_stats.
Genome chunks¶
The genome was split up into 1,371 genomic regions or 'chunks' by physical position, to process the aggregation in parallel and to ensure that the resulting output files are not so large as to make them unworkable. These chunks were determined by investigating the number of variants per chunk in aggV1 (the previous gVCF aggregation). Doing so resulted in 1,371 chunks that were comparable in size and number of variants. All 78,195 samples are in each chunk.
Chunk names
Chunks are named in the following format: gel_mainProgramme_aggV2chromosomestartstop.vcf.gz_
For example: gel_mainProgramme_aggV2chr1146620016147701894.vcf.gz_
gVCF aggregation workflow overview¶
A schematic of the aggregation process is presented in the diagram below.
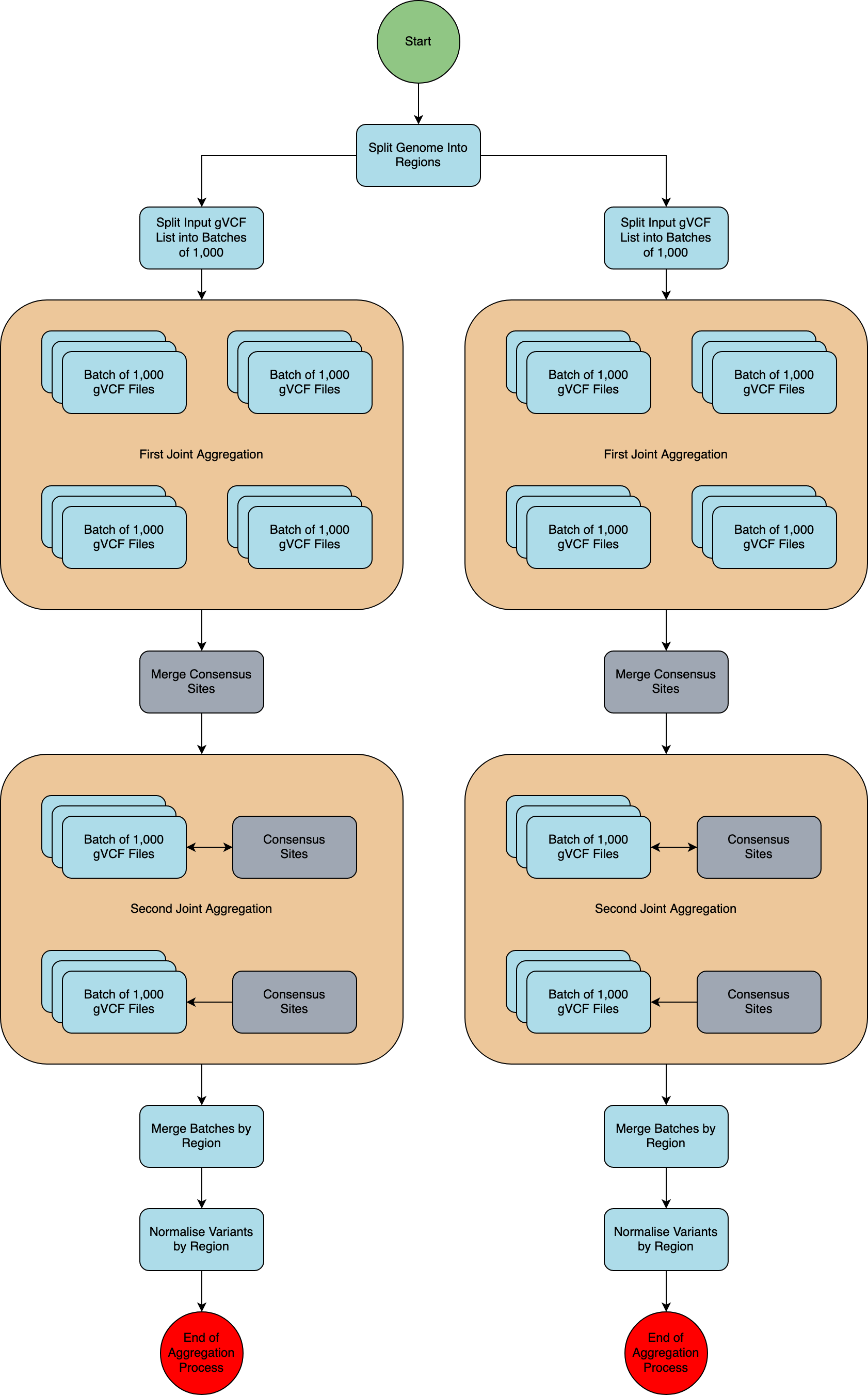
Split by genome region¶
The genome was split into 1,371 regions so that each region can be processed in parallel.
Split input into batches¶
The input file list, containing the locations of all single sample gVCF files to be included, was split into batches of 1,000 samples. This was determined to be the optimal size in terms of resource usage and compute speed for this run. Each batch of 1,000 samples was used in every region.
First joint aggregation¶
In this step, each batch of 1,000 samples is aggregated, and all the unique variants (in terms of chromosome, position, reference, and alternative alleles) for each batch are included in the output. This ensures that we capture every variant site in the cohort.
Merge consensus sites (synchronisation step)¶
The outputs of the first joint aggregation are merged to provide a final list of variants per genome region.
When splitting the input samples into batches, there will be variants in one batch that are not present in any other. The final output would therefore only contain the genotypes from the subset of the batch where it was found. The variant for all other genotypes would be set to missing, whereas most other samples will in truth carry the reference allele. The synchronisation step ensures that all variants are included for all samples, instead of only in the batch where the variant was found.
Second joint aggregation¶
Each batch of 1,000 samples is aggregated with gvcfgenotyper, and the list of variants from the Merge consensus sites step is used to force genotype each batch. This ensures that all variants are genotyped in all samples.
Merge regions¶
All batches of 1,000 samples per genomic region are merged with bcftools into a single VCF.GZ file per genomic region, resulting in a single VCF.GZ file with 78,195 samples per genomic region.
Normalise regions¶
The output of Merge regions is a VCF.gz file that contains unnormalised variants represented in their multiallelic format; where one variant can have many ALT alleles. We use the software vt to normalise and decompose all variants so that they are represented in their normalised bi-allelic format. Please see the Variant Normalisation page for full details.
Help and support¶
Please reach out via the Genomics England Service Desk for any issues related to the aggV2 aggregation or companion datasets, including "aggV2" in the title/description of your inquiry.