Small Variant workflow
The Small Variant workflow finds variants within a list of genes. It outputs these into tsv files containing variants, variant consequences and the participants with alternative alleles at these loci.
Germline genomes only
The Small Variant workflow is intended for use on germline genomes only.
Given a list of participant VCFs and a list of query genes, the workflow aggregates the VCFs into a single multi-sample VCF file keeping only variants within query genes, and annotates the variants. The default annotations include functional consequences, allele frequencies, disease associations, and pubmed citations. All included annotations are listed in the Design section. You can use the workflow to answer common queries like “Which participants have rare, missense variants in my gene(s) of interest?”
The workflow is written in Nextflow DSL2 and uses containerised Python 3 (and the Python API for LabKey), bcftools, and VEP
Sample selection
On HPC, if no samplesheet is provided, the workflow performs a LabKey query to retrieve all germline rare disease and cancer participants, for both Illumina V2 (GRCh37) and Illumina V4 (GRCh38). This retrieves that largest sample called by a single tool. To use DRAGEN (GRCh38), set parameter --dragen true (default false). On CloudOS, a samplesheet is required. The minimum sample size for each delivery version (genome build) included is four participants.
Usage¶
-
copy the LSF submission script
/gel_data_resources/workflows/rdp_small_variant/v3.1.4/submit.bsubto yourre_gecipordiscovery_forumfoldercdinto your working foldercp /gel_data_resources/workflows/rdp_small_variant/v3.1.4/submit.bsub .
-
create your gene and (optional) samplesheet input files
- update the submission script:
- insert your LSF Project Code:
#BSUB -P <PROJECT_CODE> # line 3PROJECT_CODE='<PROJECT_CODE>' # line 9
- set your input files as parameters:
--gene_input # line 41--samplesheet(optional, if using a custom sample subset)
- insert your LSF Project Code:
- run the workflow,
bsub < submit.bsub
submit.bsub
Default parameters
The default --gene-input argument is a file which includes three genes (BRCA1, TNF, ENSG00000227518). The --samplesheet parameter is null and a LabKey query returns rare disease and cancer germline samples for both Illumina V2 (GRCh37) and Illumina V4 (GRCh38). Results are written to the results/ subdirectory (change with --outdir). All workflow processes are run with containers and images are cached in your default singularity cache, $HOME/.singularity/cache.
To change workflow parameters, edit your copy of the submission script. See Parameters for additional details on querying and setting workflow parameters. See Input files for input file formats.
Tutorial video¶
Design¶
Here are the connected processes of the workflow as a directed acyclic graph (DAG).
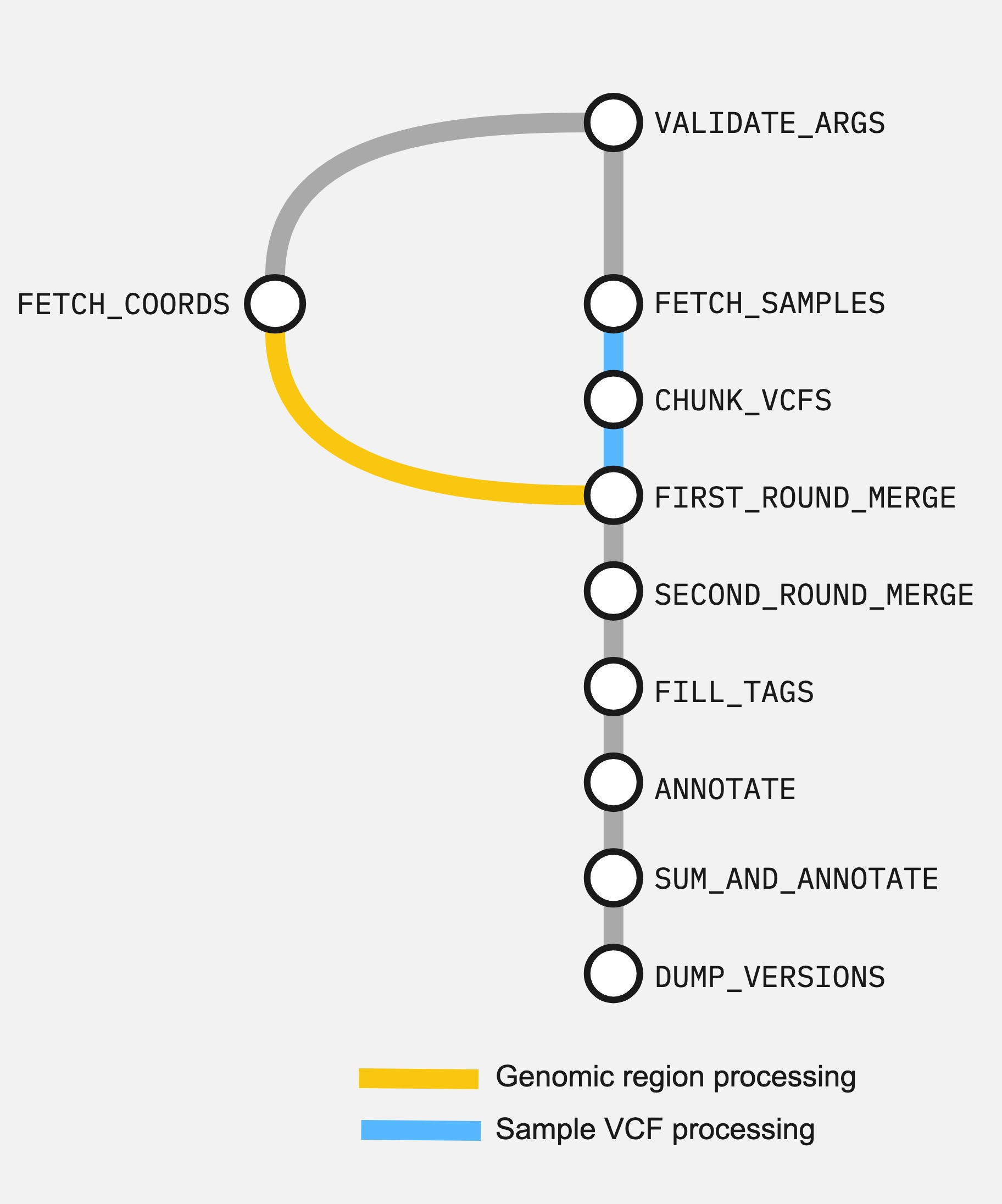
The steps are:
- Validate input arguments (
VALIDATE_ARGS) - Get coordinates for query genes (
FETCH_COORDS) - Get sample VCF list (
FETCH_SAMPLES) - Merge single-sample VCFs by chunk, selecting query genes regions (
FIRST_ROUND_MERGE) - Merge and norm to multi-sample VCF (
SECOND_ROUND_MERGE) - Compute and fill VCF INFO tags (
FILL_TAGS) - VEP-annotate variants (
VEP_ANNOTATE) - Combine +fill-tags computed INFO tags and VEP annotation (
COMBINE_ANNOTATIONS) - Write containerised tool version software_versions.yml (
DUMP_VERSIONS)
FILL_TAGS INFO tags
Computed using bcftools plugin +fill-tags:
ANTotal number of alleles in called genotypesACAllele count in genotypesAC_HomAllele counts in homozygous genotypesAC_HetAllele counts in heterozygous genotypesAC_HemiAllele counts in hemizygous genotypesNSNumber of samples with dataMAFMinor Allele frequency
VEP_ANNOTATE annotations
VEP annotations and custom ClinVar and plugin LOFTEE fields added (See Running VEP for a detailed description of options and annotations):
sift bpredicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids (both prediction term and score)ccdsadds the Consensus Coding Sequence (CCDS) transcript identifer (where available) to the outputuniprotadds best match accessions for translated protein products from three UniProt-related databases (SWISSPROT, TREMBL and UniParc)hgvsadds Human Genome Variation Society (HGVS) nomenclature based on Ensembl stable identifiers. Both coding and protein sequence names are added where appropriatesymboladds the gene symbol (e.g. HGNC) (where available)numbersadds affected exon and intron numberingdomainsadds names of overlapping protein domainsregulatorylooks for overlaps with regulatory regionscanonicaladds a flag indicating if the transcript is the canonical transcript for the geneproteinadds the Ensembl protein identifier to the output where appropriatebiotypeadds the biotype of the transcript or regulatory featuretsladds the transcript support level for this transcriptapprisadds the APPRIS isoform annotation for this transcriptgene_phenotypeindicates if the overlapped gene is associated with a phenotype, disease or traitafadds the global allele frequency (AF) from 1000 Genomes Phase 3 data for any known co-located variantaf_1kgadds allele frequency from continental populations (AFR,AMR,EAS,EUR,SAS) of 1000 Genomes Phase 3af_gnomadincludes allele frequency from Genome Aggregation Database (gnomAD) exome populationsaf_gnomadginclude allele frequency from Genome Aggregation Database (gnomAD) genome populationsmax_afreports the highest allele frequency observed in any population from 1000 genomes, ESP or gnomADpubmedreports Pubmed IDs for publications that cite existing variantvariant_classoutputs the Sequence Ontology variant classmaneadds a flag indicating if the transcript is the MANE Select or MANE Plus Clinical transcript for the gene
VEP --custom ClinVar fields: CLNDN, CLNDNINCL, CLNDISDB, CLNDISDBINCL, CLNHGVS, CLNREVSTAT, CLNSIG, CLNSIGCONF, CLNSIGINCL, CLNVC, CLNVCSO, CLNVI
VEP --plugin LOFTEE (Loss-Of-Function Transcript Effect Estimator) fields assess low- and high-confidence, stop-gained, splice site disrupting, and frameshift variants
Nextflow documentation¶
For Nextflow command line documentation use nextflow help, and nextflow <command> -help for help on a particular command. To print the workflow configuration use nextflow config -profile <profile> - configurable parameters have a params. prefix. See Parameters for use on the command line. See also Nextflow documentation and Nextflow training.