somAgg: VCF aggregation¶
Single sample annotated somatic small variant VCF files were aggregated using bcftools to create the multi-sample VCF of 16,341 samples.
The samples included in the aggregate can be found in the LabKey table cancer_analysis. These sample are already decomposed by vt and no multiallelic sites are present.
Genome chunks¶
The genome was split up into 1,371 genomic regions or chunks by physical position, to process the aggregation in parallel and to ensure that the resulting output files are not so large as to make them unworkable. These chunks were determined by investigating the number of variants per chunk in aggV1 (the previous gVCF aggregation for germline). Doing so resulted in 1,371 chunks that were comparable in size and number of variants. All 16,341 samples are in each chunk.
Chunk Names
Chunks are named in the following format: somAgg_dr12chromosomestartstop.vcf.gz_
For example: somAgg_dr12chr1146620016147701894.vcf.gz_
Somatic VCF aggregation workflow overview¶
A schematic of the aggregation process is presented in the diagram below.
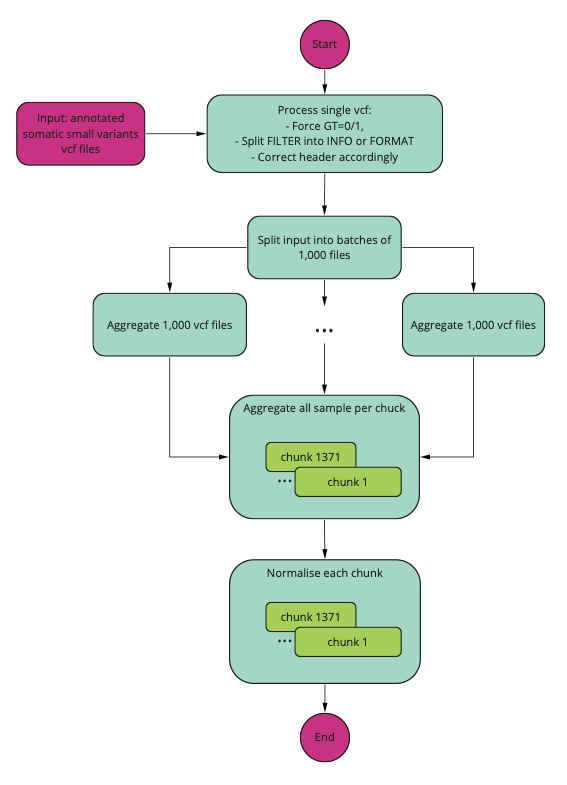
Pre-processing of single VCF files¶
Each single VCF file had GT forced into 0/1 for all called variants. Also FILTER flags have been split into INFO and FORMAT. The former received flags for site, such as commonGermlineVariant or ReceptiveRegion, while the later those related to sample, indication that the reads supporting the call did not have strong evidence.
Split input into batches¶
The input file list, containing the locations of all single sample annotated somatic VCF files to be included, was split into batches of 1,000 samples. This was determined to be the optimal size in terms of resource usage and compute speed for this run. Each batch of 1,000 samples was used in every region.
Batch aggregation¶
Each 1000 VCF files was aggregated using bcftools merge. Missing GT were forced to 0/0 and no new multiallelic was allowed since the input VCF files had already being decomposed.
Split of genome into chunks and all sample aggregation¶
The genome was split into 1,371 regions so that each final VCF files has a manageable size.
All 1000 VCF file batches were aggregated using bcftools merge. Missing GT were forced to 0/0 and no new multiallelic was allowed since the input VCF files had already being decomposed.
Normalise regions¶
The aggregated vcf files had been normalised to assure indels were represented in an a unique away after the aggregation. A normalised variable is
- parsimonious, i.e. is represented in as few nucleotides as possible without an allele of length 0, and
- left-aligned, i.e. it is not possible to shift its position to the left while keeping the length of all its alleles constant.
Normalisation was performed by bcftools norm.
Variants representation¶
The resulting aggregate chuck of the somAgg contains variants represented in bi-allelic format and have genotype 0/1 if observed in a sample. The bi-allelic format is assured by the input single sample VCF files, which are already decomposed and contain all variants in bi-allelic format. During the bcftools merge steps no multiallelic are allowed. Note:
- Multi-allelic: where a single variant contains three or more observed alleles, counting the reference as one, therefore allowing for two or more variant alleles (heterozygous genotype example: 1/2 – note there are none in somAgg)
- Bi-allelic: where a variant contains two observed alleles, counting the reference as one, and therefore allowing for one variant allele (genotypes are always: 0/1)
In addition, all variants that are not observed in a sample have genotype 0/0.
Variants that were multiallelic in the single VCF files are annotated with SAMPLE_MULTIALLELIC (for SNVs) and SAMPLE_VARIANT (for indels) in the FORMAT field.
Help and support¶
Please reach out via the Genomics England Service Desk for any issues related to the somAgg aggregation or companion datasets, including "somAgg" in the title/description of your inquiry.