100kGP Release v18 (21/12/2023)¶
This document provides a description of the 100k Genomes Project (previously known as Main Programme) Data Release v18 dated 21st December 2023.
Each progressive release incorporates new content, enhances existing content, and enables more effective use of the data in the National Genomics Research Library (NGRL).
This data are presented within the Genomics England Research Environment, accessed via the AWS virtual desktop interface and subject to all Genomics England data protection and privacy principles.
Please see the Research Environment User Guide for detailed documentation on how to use and query the Genomics England dataset.
Genomes in this release¶
Data Release Version 18 provides clinical data for 90,178 participants, and 106,263 genomes (uniquely sequenced samples) from 88,492 of these participants. We hold germline genomes from 72,869 rare disease programme participants1 and germline and somatic genomes from 15,623 cancer programme participants2 from the 100k Genomes Project.

Table containing genome counts by sample, and delivery types.3
| Programme | Sample Type | Delivery Type | Genomes | Participants |
|---|---|---|---|---|
| Cancer | Germline | V2 (GRCh37) | 265 | 265 |
| Somatic | V2 (GRCh37) | 294 | 265 | |
| Cancer | Germline | V4 (GRCh38) | 15,772 | 15,422 |
| Somatic | V4 (GRCh38) | 17,197 | 15,615 | |
| Cancer | Germline | D2 (GRCh38) | 15,756 | 14,999 |
| Somatic | D2 (GRCh38) | 16,048 | 15,000 | |
| Cancer | Germline | Combined | 31,793 | 15,618 |
| Somatic | Combined | 33,539 | 15,617 | |
| Cancer | Total | Combined | 65,3324 | 15,623 |
| Rare Disease | Germline | V2 (GRCh37) | 10,336 | 10,249 |
| Germline | V4 (GRCh38) | 65,583 | 64,894 | |
| Rare Disease | Total | Combined | 75,919 | 72,869 |
| Total | Combined | Combined | 141,2515 | 88,492 |
The above table contains genomes that have been processed using different alignment methods and therefore can be counted multiple times. The table below provides the number of uniquely sequenced samples.
| Programme | Sample Type | Genomes count | Participant count |
|---|---|---|---|
| Cancer | Germline | 15,901 | 15,618 |
| Tumour | 17,002 | 15,617 | |
| Cancer | Total | 32,751 | 15,623 |
| Rare Disease | Germline | 73,512 | 72,869 |
| Total | 106,263 | 88,492 |
Clinical data in this release¶
Bug in rtds table
There is a bug in the NCRAS radiotherapy table, rtds. Approximately 8% of all records in this table are missing dates. This is due to a bug translating Sep in three-letter months to numbered dates.
The Genomics England 100k Genomes Project clinical data are organised into tables found in LabKey. You can find details of these tables and their contents in our clinical data documentation, and data dictionary.
Activity period coverage for the longitudinal secondary data tables¶
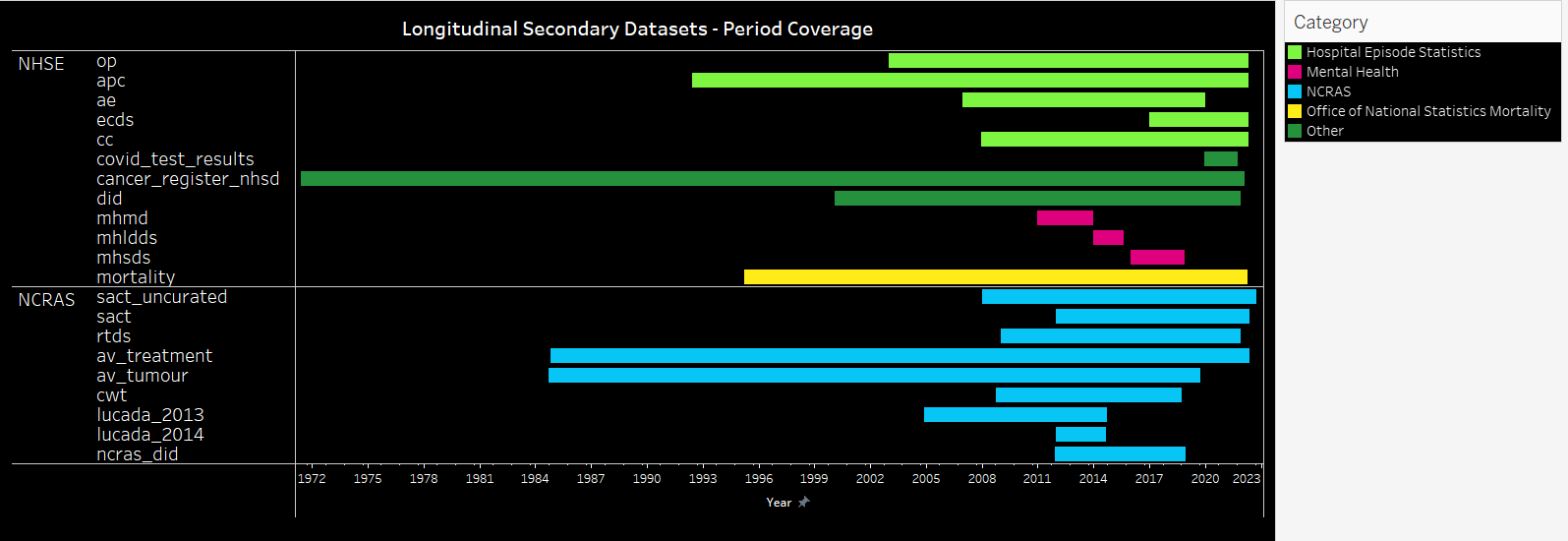
| Source | Category | Dataset | Start | End |
|---|---|---|---|---|
| NHSE | Hospital Episode Statistics | op | 01/04/2003 | 31/07/2022 |
| NHSE | Hospital Episode Statistics | apc | 13/09/1992 | 31/07/2022 |
| NHSE | Hospital Episode Statistics | ae | 01/04/2007 | 31/03/2020 |
| NHSE | Hospital Episode Statistics | ecds | 05/04/2017 | 04/08/2022 |
| NHSE | Hospital Episode Statistics | cc | 01/04/2008 | 29/07/2022 |
| NHSE | Other | covid_test_results | 16/03/2020 | 05/01/2022 |
| NHSE | Other | cancer_register_nhsd | 07/09/1971 | 15/05/2022 |
| NHSE | Other | did | 19/05/2000 | 28/02/2022 |
| NHSE | Mental Health | mhmd | 01/04/2011 | 31/03/2014 |
| NHSE | Mental Health | mhldds | 01/04/2014 | 30/11/2015 |
| NHSE | Mental Health | mhsds | 01/04/2016 | 01/03/2019 |
| NHSE | Office of National Statistics Mortality | mortality | 27/06/1995 | 19/07/2022 |
| NCRAS | NCRAS | sact_uncurated | 09/04/2008 | 30/12/2022 |
| NCRAS | NCRAS | sact | 06/04/2012 | 30/08/2022 |
| NCRAS | NCRAS | rtds | 15/04/2009 | 28/02/2022 |
| NCRAS | NCRAS | av_treatment | 08/02/1985 | 22/08/2022 |
| NCRAS | NCRAS | av_tumour | 01/01/1985 | 31/12/2019 |
| NCRAS | NCRAS | cwt | 05/01/2009 | 31/12/2018 |
| NCRAS | NCRAS | lucada_2013 | 25/02/2005 | 02/01/2015 |
| NCRAS | NCRAS | lucada_2014 | 23/03/2012 | 08/12/2014 |
| NCRAS | NCRAS | ncras_did | 01/03/2012 | 26/03/2019 |
Change summary¶
Updated 18/03/2024 Transcriptomics pilot data, (transcriptome_file_paths_and_types, rnaseq_qc_metrics)¶
Two new tables have been added to the release as part of the additional transcriptomics pilot data beign included in the release. Further details on these datasets can be found in the Data Dictionary provided.
cancer_ont_cohorts¶
The table now includes information about BAM files that contain methylation tags for 9 participants. Six new columns have been added to record the versions of the tools used to call methylation sites plus a column with the file paths:
methylation_guppy_version: Guppy toolkit version used for methylation BAMs.methylation_basecall_version: Guppy basecalling version used for methylation BAMs.methylation_basecall_model: Model used for guppy basecalling for methylation BAMs.methylation_basecall_filter_threshold: Basecalling filter threshold value used for methylation BAMs.methylation_minimap2_version: Minimap2 version used for read alignment for methylation BAMs.lr_merged_tumour_methylation_path: Path of the long read alignment data with methylation tags for the tumour sample.
submitted_diagnostic_discovery¶
The table now includes two new columns:
group: Combination of the participant id and the gene name used to group compound heterozygous variants together.Zygosity: Whether the variant is homoygous, heterozygous, compound heterozygous, heterozygous or mosaic.
In addition, the gene_build column has been renamed to genome_build, which is the nomenclature we use in other tables such as sequencing_report and genome_file_paths_and_types.
domain_assignment¶
Table has been decommissioned.
Changes video¶
How to find release 18 data¶
LabKey¶
The clinical data, secondary data, and tabulated bioinformatic data for this data release, and the paths to the applicable genome files, are found in the following LabKey folder:
main_programme/main-programme_v18_2023-12-21
Flatfiles¶
Genomics England data resources are available in the following locations:
From the AWS desktop:
~/gel_data_resources/
From the high performance compute (HPC) cluster:
/gel_data_resources/
The data resources available here are:
| Data | Format | Associated LabKey tables |
|---|---|---|
| Genome alignments | BAM or CRAM | genome_file_paths_and_types |
| Variant calls | VCF | genome_file_paths_and_types |
| Cancer tiering reports | JSON | cancer_tier_and_domain_variants |
| Structural and copy number variant reports | JSON | n/a |
| Aggregated gVCF dataset (aggV2) | aggregated VCF | aggregate_gvcf_sample_stats |
Scope¶
In scope¶
Data that are in scope for this release:
- Cancer and rare disease data for the main programme participants with current consent. These data include:
- Genomic data for participants when available
- Whole genome sequencing (WGS) family-based quality control for rare disease, reporting sex checks and pedigree checks
- Outputs of the Genomics England Bioinformatics Research Services
- An aggregated Illumina gVCF for germline genomes (including genomes up to late 2019). Please see the documentation here: Aggregated Variant Calls (aggV2)
- Principal components for germline genomes
- Inferred ancestry assigned to samples based on genomic data. (See also aggV2)
- The list of samples for this aggregate can be found in the 'aggregate_gvcf_sample_stats' table in Labkey, for the latest data release.
- Phased data is now available for participants in our aggV2 dataset. The data contains over 342 million phased small variants (SNPs and short indels) across chromosomes 1-22 of aggV2. Detailed documentation can be found here: Aggv2 Phased Data (Provided by University of Oxford). This data was provided by Sinan Shi from the University of Oxford.
- A somatic aggregate containing 16,341 somatic vcf files (including genomes up to early 2021) from the 100,000 Genomes Project which we made available as a multi-sample VCF dataset (somAgg). More information can be found here: Somatic Aggregated Variant Call (somAgg v0.2 ALPHA version). This is an early stage release and feedback is very welcome.
- Annotated single nucleotide variants and small indels (≤50bp) from quality controlled tumour whole genomes.
- Genome-wide de novo variant dataset for 13,917 trios from 12,577 families from the rare disease programme. This was built for main programme data release v9. Researchers are responsible for only using the participants who are consented for research in the latest data release. Please see the documentation here: The de novo variant research dataset for the 100,000 Genomes Project
- Polygenic Risk Score values have been made available for 12 complex traits, for ~40k participants from the aggV2 dataset. Detailed documentation can be found here: Polygenic Risk Scores (Provided by Genomics PLC). This data was provided by Genomics PLC.
- A collection of 18,990 cancer tier and domain variants reported through the bioinformatics cancer interpretation pipeline aggregated in the cancer_tier_and_domain_variants table.
- Long read sequencing data provided by internal teams as part of collaborative pilot projects. These are now been placed under a separate section in LabKey called 'Long Read Sequencing'.
- Oxford Nanopore Technologies (ONT)
lrs_laboratory_sampleandlrs_sequencing_data: <100 Rare Disease programme participants.cancer_ont_cohorts: 101 Cancer programme participants from various cancer disease types. Starting in V18, we also include BAM files with methylation sites called.
- PacBio
rare_disease_pacbio_pilot: <100 Rare Disease programme participants
- Oxford Nanopore Technologies (ONT)
- Outputs of the Genomics England Bioinformatics rare diseases interpretation pipeline
- Tiering data – rare disease
- Exomiser results for interpreted genomes – rare disease
- GMC outcome data ("exit questionnaire data") – rare disease - up until 02/12/2023.
- Platypus vcfs used for the genomic interpretation of 33,966 families (3,430 GRCh37 and 30,538 GRCh38) are available via the rare_disease_interpreted table. Platypus vcfs are provided unannotated.
- Outputs of the Genomics England Bioinformatics cancer interpretation pipeline
- 'Gold standard' cancer genomes which have been through interpretation and passed quality checks
- Tumour signature and mutational burden data
- Annotation and tiering of small variants
- Tiering, structural and copy number variant report
- Cancer Principal Component Analysis (PCA). For more information on these metrics please see the following document: Cancer Analysis Technical Information Document.
- Submitted Diagnostic Discovery data provided by the Research Community. These are potential diagnoses that were not identified as causal in the initial analysis by GEL, but were identified by Research Environment users and submitted to GEL as part of a process called 'Diagnostic Discovery'. If you would like to be involved in this yourself please contact service desk. These findings are in addition to the GMC exit questionnaire data, and may remain listed as "unsolved cases" therein.
- Clinical data collected upon enrollment, including formal pedigree data on rare disease participants where it is available
- Secondary datasets (medical history), these are available at varying levels of completeness and include:
- Hospital Episode Statistics (HES), including HES Accident and Emergency, HES Admitted Patient Care, and HES Outpatient Care.
- Emergency Care Data Set (ECDS).
- Diagnostic Imaging Dataset (DID).
- Mental Health Minimum Dataset (MHMDS).
- Mental Health Learning Disabilities Dataset (MHLDDS).
- Mental Health Services Dataset (MHSDS).
- Office for National Statistics - Death details data, Cancer Registration (MORTALITY, CANCER_REGISTRY).
- Systemic Anti-Cancer Therapy Dataset (SACT).
- Systemic Anti-Cancer Therapy Dataset - UNCURATED (SACT_UNCURATED).
- National Radiotherapy Dataset (RTDS).
- Cancer Registration (AV) tables.
- Cancer waiting times (CWT).
- Lung Cancer Data Audit (LUCADA).
- National Cancer Registration and Analysis Service Diagnostic Imaging Dataset (NCRAS_DID).
- COVID Test Results data (covid_test_results). This was previously included in the frequent release section.
- Sample datasets describing:
- Handling and quality control of DNA samples at the Genomic Medicine Centres, the biorepository and the sequencer.
- Omics samples stored at the biorepository.
- Orthogonal standard-of-care test data collected from GMCs for a subset of cancer patients
- Updated 18/03/2024 Transcriptomics (RNA-sequencing) Pilot data provided by internal teams as part of collaborative pilot projects. These are now been placed under two tables in Labkey called 'transcriptome_file_paths_and_types' and 'rnaseq_qc_metrics'
- The GEL Transcriptomics Pilot comprises RNA-sequencing of a subset (>5,000) of rare disease probands from the 100,000 Genomes Project who did not receive a genetic diagnosis through the Genomics England Interpretation Pipeline. We prioritised probands who were found to carry variants of unknown significance.
Out of scope¶
Additional time is required to update the applications/tools that are available in the RE to the current data release, e.g. IVA, Participant Explorer. Please refer to the Application Data Versions page for the data release version used in the RE products and services.
Data out of scope for this release:
- Clinical and genomic data for participants that have withdrawn from the 100,000 Genomes Project or were otherwise ineligible.
- Participant data from the pilot phases of the project.
- Clinical data for participants on expired child consent collected after their 16th birthday (for more details see Clinical and phenotype data : Secondary data - Participant Consent).
- Data relating to the NHS Genomic Medicine Service (GMS) . Genomic and clinical data on NHS GMS participants is currently released separately to the main_programme release. For more information on the NHS GMS data releases please see: NHS GMS data release notes
Quality notes¶
- BAM and VCF genomic data files are as they have been delivered to us by our sequencing provider (Illumina). These have all passed an initial QC check based on sequencing quality and coverage. They have, however, not all undergone our full in-house quality checks and they are therefore subject to potential discrepancies or inaccuracies. Such checks include, but are not limited to, discrepancies in genetic versus reported sex and in family relationships.
- As participants undergo the in-house checks and pass through the Genomics England interpretation pipeline, any inaccuracies we identify will be rectified in subsequent releases.
- Any samples that have been affected prior to this release (e.g. sample swaps or samples that have been retracted as part of the in-house QC process) are listed in Section 10 below.
- You are encouraged to work on the subset of samples that have already passed our internal QC checks; these can be found below for rare disease and cancer genomes, respectively.
- For Rare Disease genomes, you should note that all tiered genomes have passed through Genomics England in-house QCs and that all tiered genomes come from the pool of genomes that have had family checks applied to them, as a first step towards Genomics England tiering. For rare disease interpretation including tiering, small variants are called using the Platypus variant caller. Please see the Rare Disease Results Guide on our Further reading and documentation page for more information.
- Different QC filtering has been applied to the Illumina VCF files and the Platypus VCFs that are used for tiering in the rare disease programme. There may therefore, be tiered variants that have been filtered out of the Illumina VCF files, and, conversely, variants present in the Illumina VCF file that have been filtered out of the platypus VCFs.
- Some rare disease families lack a proband.
- Human Phenotype Ontology (HPO) terms may be missing or incomplete for some participants.
- Each participant's relationship to their family's proband is available in the
rare_diseases_pedigree_membertable and can be used to determine family relationships, especially for cases without formal pedigree data. Pedigree data are only available for a subset of rare disease participants. - WGS family selection quality checks are provided for rare disease genomes on GRCh38, reporting abnormalities of sex chromosomes and reported vs genetic sex summary checks (computed from family relatedness, Mendelian inconsistencies, and sex chromosome checks). Full details on why a family has failed a reported vs genetic sex check can be requested via the Service Desk.
- For Cancer genomes, you should note that all 'gold standard genomes' that have been through Genomics England interpretation and passed quality checks are found in the cancer quick view table cancer_analysis. We strongly recommend using the data from this table for all cancer analyses.
- Clinical data and secondary data have been provided as submitted and have undergone limited validation and cleaning
- sact_uncurated is the table with the raw data from NCRAS which feeds into their curation process producing the SACT table, which remains the gold standard. A major point to raise is that neither of the SACT tables contain tumour IDs, thus you must match this dataset to other NCRAS registries by adjusting for date. A lot of familiar data fields remain in their raw non-standardised form (sex, treatmentintent, clinicaltrialindicator). Pending feedback, these fields can be normalised in subsequent updates.
Terms of use for specific cohorts¶
Participants identified as TracerX in the field normalised_consent_form in the participant table in LabKey must not be used by commercial organisations. Commercial organisations do not have access to the genomic data of TracerX participants.
Participants with a participant ID that commences with 125 or 226 were recruited through the Scottish Genomes Partnership Research Programme. These are under the governance of a separate but linked consent and protocol to the 100,000 genomes project. Only the removal of summary level statistics is permitted. Airlock approval will not be granted for the removal of record level data associated with these participants.
Cohort metadata¶
Within the data release, there is genomic data and clinical data for participants that are part of non-NHS research cohorts that have been sequenced by Illumina and analysed via the Genomics England pipeline.
These research cohorts can be distinguished via their clinic ID as each has been given their own unique code. These clinic IDs are primarily located in the participant table filtering either the registered_at_ldp_ods_code or registered_at_ldp_bioinformatics_ods_code for the respective clinic ID. If any genomic or clinical data from the research cohorts is used in your analysis and subsequent publication, reference to the cohort organisation in the first column of the below table will need to be made.
| Non-NHS Cohort Name | Clinic ID | Rare Disease/ Cancer | Description | Constraints | Requirements of Use | Number of genomes (in v18 data release) | Opportunities for further research |
|---|---|---|---|---|---|---|---|
| Breast Cancer Now | BCN | Cancer | The Breast Cancer Now Tissue Bank (BCNTB) is a multi-centre tissue bank established to fill the gap in the Triple Negative breast cancer (TNBC) research community. It systematically collects high quality tissues and data under an established ethical framework. Full clinico-pathological and follow-up data is due to be made available with ongoing longitudinal data collection. This cohort is curated group of 81 treatment naïve TNBC patients. Additional tissue for many is available through the BCNTB for further matched ‘omic analysis. |
Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | 81 | Potential to remove identifiers for the purpose of requesting access to Breast Cancer Now biobank samples. |
| Genomics England CLL pilot study | CLL | Cancer | The original Chronic lymphocytic leukaemia (CLL) Genomics England Pilot aimed to develop the protocols and analytical methods required to perform whole genome sequencing (WGS) at scale for patients with CLL recruited into national clinical trials as a prelude to the Genomics England main programme. This cohort is a small subset of the pilot to allow for the provision of validation data. | Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | 4 | |
| UKALL2003 trial | ALL | Cancer | The aim of this project is to explore the genomic landscape of patients with acute lymphoblastic leukaemia at initial presentation in order identify mutations that could explain their poor response and potentially be future biomarkers. The objective was to perform whole genome sequencing and targeted screening for mismatch repair deficiency on a large well annotated cohort of patients with ALL treated on the UKALL2003 trial. This will generate, for the first time, a comprehensive genomic landscape of chemo-resistant acute lymphoblastic leukaemia. | Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | 67 | |
| NIHR Bioresource | NB3 | Rare Disease | The NIHR BioResource is comprised of volunteers from around the country who have given their consent to taking of a biological sample, and they are willing to be approached to participate in research studies and trials on the basis of their genotype, and or phenotype. This cohort consists of rare disease participants who consented to WGS as part of the 100,000 Genomes Project. | Consistent with Genomics England acceptable uses, | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | 309 | Anyone who wishes to be granted permission to contact any of the NIHR BioResource participants should follow the process of applying to the NIHR BioResource. The steps to be made can be found on the NIHR BioResource website |
Contact and support¶
For all queries relating to this data release please contact the Genomics England Service Desk portal: Service Desk (accessible from outside the Research Environment). The Service Desk is supported by dedicated Genomics England staff for all relevant questions.
-
Some Rare Disease participants have multiple genomes, aligned to both GRCh37 and GRCh38 ↩
-
This excludes 86 TracerX genomes from 99 participants (refer to 6.4 for further information). ↩
-
Long read samples are excluded from these counts. ↩
-
On average 2.1 genomes per cancer participant per delivery type. ↩
-
These counts exclude experimental types. ↩