Structural Variant workflow¶
The Structural Variant workflow allows you to find all structural variants in a list of genes or genomic regions.
The workflow (previously the SV/CNV workflow) extracts canonical structural variants (insertions, deletions, duplications, inversions, translocations) and copy number variants within query regions defined by a list of genes or coordinates, from Illumina Structural Variant VCF files for germline and/or somatic cohorts. The main output is a list of observed Manta-called SVs and Canvas-called CNVs within query regions. CNVs on sex chromosomes depend on sex chromosome karyotype and should be interpreted with caution.
The workflow is written in Nextflow DSL2 and uses containerised R (and the RLabKey API), Python 3, bcftools, and bedtools.
Note
In the RE (HPC), if no participant VCF list is provided, the workflow performs a LabKey query to retrieve a list of standard Illumina VCF file paths for all germline rare disease and cancer participants. The default selection drops DRAGEN-called genomes, in order to get the largest sample called by a single tool. You can use alternate VCF files (e.g. platypus, DRAGEN) by supplying a VCF list. In CloudRE (CloudOS), a participant VCF list is required.
Design¶
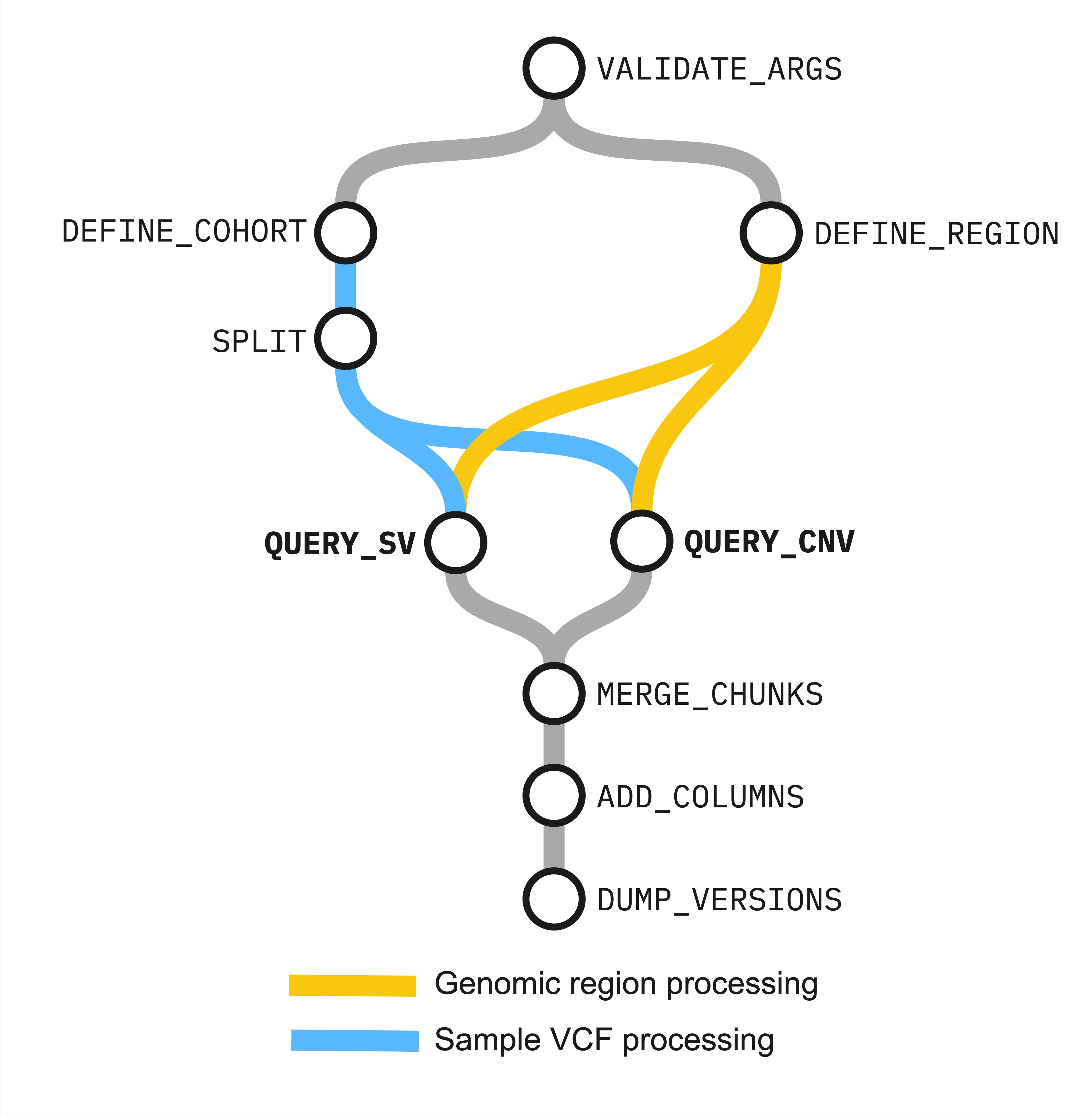
Figure. Relationship between workflow processes in Structural Variant 3.0
Processes¶
- check the validity of parameter arguments (
VALIDATE_ARGS) - get coordinates and region information (
DEFINE_REGION) - get a list of VCF files for each cohort (
DEFINE_COHORT) - chunk VCF lists (
SPLIT) - query chunked VCFs for MANTA-called structural variants(
QUERY_SV) - query chunked VCFs for CANVAS-called copy-number variants (
QUERY_CNV) - merge chunked variant outputs (
MERGE_CHUNKS) - add variant query region overlap information (
ADD_COLUMNS) - log containerised tool versions (
DUMP_VERSIONS)
Usage¶
Warning
Before running the workflow in the RE (HPC), you must configure your R LabKey API access.
Using the submission script¶
Navigate to your re_gecip or discovery_forum folder, create a directory for your analysis, e.g. my_analysis, and copy the submission script
Copy the submission script only, not the entire directory.
The submission script simply loads the required Nextflow and singularity modules and uses the Nextflow command line tool to run the workflow.
- replace
<project_code>with your LSF Project Code. -
replace
<path_to_input_file>with a path to an input file, i.e., either a one gene per line file for--input_type 'gene', or a bed file of regions for--input_type 'region'. Create this input file in your working directory, e.g., my_analysis/. See example input files here:- /pgen_int_data_resources/workflows/rdp_structural_variant/v3.1.2/input/genes.txt for --input_type 'gene', or
- /pgen_int_data_resources/workflows/rdp_structural_variant/v3.1.2/input/regions.bed for --input_type 'region'
#!/bin/bash #BSUB -P <project_code> #BSUB -q inter #BSUB -R rusage[mem=10000] #BSUB -M 12000 #BSUB -J strv #BSUB -o logs/%J_strv.stdout #BSUB -e logs/%J_strv.stderr LSF_JOB_ID=${LSB_JOBID:-default} export NXF_LOG_FILE="logs/${LSF_JOB_ID}_strv.log" export NXF_DISABLE_CHECK_LATEST=true module purge module load singularity/4.1.1 nextflow/23.10 mkdir -p ./logs/ structural_variant='/pgen_int_data_resources/workflows/rdp_structural_variant/v3.1.2' nextflow run "${structural_variant}"/main.nf \ --project_code '<project_code>' \ --data_version 'main-programme_v18_2023-12-21' \ --input_file '<path_to_input_file>' \ --input_type 'gene' \ --sample_type 'all' \ --genome_build 'both' \ -profile cluster \ -ansi-log false \ -resume -
run the workflow on the default LabKey query sample.
In the RE (HPC), the default sample (if no sample file is supplied) is derived from a LabKey query based on the arguments to --sample_type, --genome_build, and --dragen. The default LabKey query draws data from two tables: genome_file_paths_and_types and participant. For reference, the full LabKey SQL query is included below. The workflow further filters this query based on --sample_type, --genome_build, and --dragen. In particular, given a participant_id is associated with 1 or more platekeys, we select the latest delivery/genome, grouping on delivery_version, cohort_type, variation type (CNV, SV), and participant_id.
define_cohort.sql
SELECT DISTINCT
g.participant_id,
CAST(g.delivery_date AS TIMESTAMP) AS date_timestamp,
g.delivery_version,
g.genome_build,
g.type AS cohort_type,
g.file_path AS sv_vcf_filepath
FROM
genome_file_paths_and_types g
INNER JOIN
participant p ON p.participant_id = g.participant_id
WHERE
LOWER(g.file_sub_type) = 'structural vcf'
AND (
g.type IN ('rare disease germline') OR
(g.type IN ('cancer germline', 'cancer somatic') AND NOT LOWER(g.delivery_version)='v2')
)
AND
LOWER(g.file_path) NOT LIKE '%itd.vcf.gz'
AND
LOWER(p.programme_consent_status) = 'consenting'
Tutorial video¶
Input¶
Below are the few input parameters you need to edit to define your query. See Parameters for the full list of pipeline parameters and their use on the command line.
--project_code LSF Project Code
--input_type'gene'|'region'- Retrieve structural variants within regions defined by
'gene'(s) or'region'(s). Default'gene'. --input_file- For
input_type = 'gene', a path to a one-per-line text file of query genes which can include both HGNC symbols and Ensembl IDs. Forinput_type = 'region', a path to a headless four-column tab-delimited bed file (chr, start, stop, name) of query regions. Default'/pgen_int_data_resources/workflows/rdp_structural_variant/main/input/genes.txt', for defaultinput_type = 'gene'. --sample_type'all'|'ca-all'|'germline'|'ca-germline'|'rd-germline'|'ca-somatic'-
- Select structural VCF sample type. This parameter is used to define the LabKey query when no sample file is supplied. Default
'all'. 'all': A combination of germline and somatic, rare disease and cancer genomes'ca-all': A combination of germline and somatic, cancer genomes only'germline': Germline only, rare disease and cancer genomes'ca-germline': Germline only, cancer genomes only'rd-germline': Germline only, rare disease genomes only'ca-somatic': Somatic only, cancer genomes only
- Select structural VCF sample type. This parameter is used to define the LabKey query when no sample file is supplied. Default
--sample_file- Either
null, or a path to a tab-delimited sample file with header:cohort_type(germlineorsomatic),genome_build(GRCh37,GRCh38orboth)sv_vcf_filepath. Defaultnull, performs a LabKey query to collect a list of structural VCFs. --data_version- This parameter is used to define the LabKey query when no sample file is supplied. Default
'main-programme_v17_2023-03-30'. --outdir- A path to an output directory. Default
'results', writes all output to a directory called 'results' in the current working directory. --genome_build'GRCh38'|'GRCh37'|'both'- This parameter is used to define the LabKey query when no sample file is supplied. Default
'both'. --dragen- Boolean indicating whether query samples include dragen-aligned VCFs. This parameter is used to define the LabKey query when no sample file is supplied. Default
false.
Parameters¶
Default workflow parameters defined in the configuration files, can be overridden on the command line. For example, to use coordinates to specify a region(s) of interest defined in a bed file, change input_type to region and supply a bed file to input_file.
nextflow run "${structural_variant}"/main.nf \
--input_file "${structural_variant}/input/regions.bed" \
--input_type 'region' \
--sample_type 'all' \
--genome_build 'both' \
-profile cluster \
-resume
Parameters and options
On the command line, prefix workflow parameters with --; prefix Nextflow options with -.
For a complete list of workflow parameters (configurable parameters have a params. prefix).
structural_variant='/pgen_int_data_resources/workflows/rdp_structural_variant/main'
nextflow config -flat -profile cluster "${structural_variant}"/main.nf | grep params.
Output¶
| file | content |
|---|---|
| SVmergedResult_chr<N>_<gene>_<ensembl>_<build>_<cohort>.tsv | Manta-called SVs observed in query regions |
| CNVmergedResult_chr<N>_<gene>_<ensembl>_<build>_<cohort>.tsv | Canvas-called CNVs observed in query regions |
| <build>_adjusted_input.tsv | Query genes/regions with HGNC and Ensembl IDs, chromosome, coordinates, etc. |
| inaccessible_vcf.tsv | VCFs for all queries and genome builds that could not be accessed |
| pipeline_info/ | Directory containing timestamped pipeline run information (execution_report_*.html, execution_timeline_*.html, execution_trace_*.txt, pipeline_dag_*.html) |
A tab-delimited text file with a header line and the following columns:
| column | description |
|---|---|
| SAMPLE | Participant platekey ID |
| CHROM | Chromosome |
| POS | Start position of the variant |
| ID | Variant ID |
| REF | Reference allele |
| ALT | Alternate allele |
| QUAL | Quality |
| FILTER | Filter applied to data |
| INFO/SVTYPE | Type of structural variant: duplication (DUP), deletion (DEL), inversion (INV), insertion (INS) or translocation (BND) |
| INFO/SVLEN | Variant size |
| INFO/END | End/Continuation position of the variant |
| GT | Genotype (0/1 = heterozygote, 1/1 = homozygote) - germline |
| GQ | Genotype quality - germline |
| PR | Spanning paired-read support for the ref and alt alleles in the order listed |
| SR | Split reads for the ref and alt alleles in the order listed, for reads where P(allele|read) > 0.999 |
| OVERLAP | 'PARTIAL' or 'TOTAL' overlap of the query region, by the variant |
| OVERLAP_CNT | Count of query region base pairs overlapped by the variant |
| OVERLAP_PCT | Percentage of query region overlapped by the variant |
A tab-delimited text file with a header line and the following columns (depedent on dragen=true or false, cnv_pass_only=true or false, germline or somatic):
| column | description |
|---|---|
| SAMPLE | Participant platekey ID |
| CHROM | Chromosome |
| POS | Start position of the variant |
| ID | Variant ID |
| REF | Reference allele |
| ALT | Alternate allele |
| FILTER | Filter applied to data |
| INFO/END | End position of the variant |
| INFO/SVTYPE | CNV for variants with copy number != 2. LOH for variants with copy number = 2, and loss of heterozygosity |
| CN | Copy number |
| GT | Genotype - 'germline', 'germline' + dragen, 'somatic' + dragen |
| MCC | Major chromosome count (equal to copy number for LOH regions) - except 'germline' + dragen or 'germline' + 'GRCh37' |
| OVERLAP | 'PARTIAL' or 'TOTAL' overlap of the query region, by the variant |
| OVERLAP_CNT | Count of query region base pairs overlapped by the variant |
| OVERLAP_PCT | Percentage of query region overlapped by the variant |
A tab-delimited text file with a header line and the following columns (first four columns only for input_type = 'region', where column four values are region identifiers):
| column | description |
|---|---|
| chr | chromosome |
| start | start position of gene region |
| end | end position of gene region |
| gene | HGNC gene symbol |
| ensemblID | Ensembl gene ID |
| internalID | identifier generated internally which will be used as a prefix for output files: chr_gene_ensemblID |
| posTag | position tag (chromosome_start_end) aiding the identifier deduplication step |
| originalInput | specifies what has been used as an input by you, either "gene" or "ensembl", referring to either gene symbol or Ensembl ID |
| entryDuplicate | NA, or containing a comma-separated string of internalIDs. The internalIDs listed here are considered the same entry due to their identical genomic position. |
Nextflow documentation
For Nextflow command line documentation use nextflow help, and nextflow <command> -help for help on a particular command. See Nextflow documentation and Nextflow training.