IVA variant browser¶
The IVA variant browser allows you to search all variants in the selected 100kGP dataset by filters such as genes, consequences and frequency.
To use the Variant browser, open IVA, Login and select a study then click Variant Browser on the top menu.
This will take you to a browser listing all the variants found in participants in that study. These are listed by their genomic location, and will include the variant identifier if this is available.
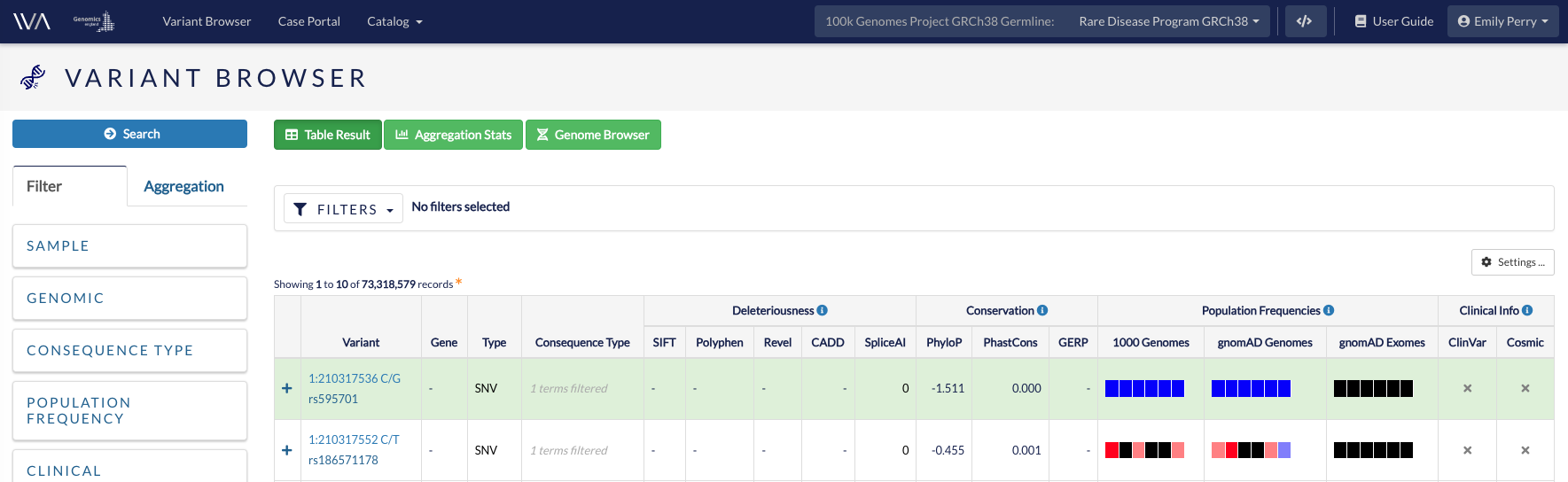
Applying filters¶
You can filter the variants in the table to find variants of interest.
The filters currently available are:
- Sample and Cohort
- Search for up to three participants.
- Alternate allele frequency in the cohort (study)
- Genomic
- Variant IDs
- Chromosomal location (by genomic coordinate)
- Feature IDs (genes, SNPs, transcripts)
- Biotype (gene or transcript type)
- Variant type - SNV (single nucleotide variant), INDEL (< 50 base pairs), INSERTION (> 50 base pairs), DELETION (> 50 base pairs), MNV (multiple nucleotide variant), SV (structural variant)
- Consequence type
- Consequence type SO (Sequence ontology) terms. You can filter for all Loss-of-Function terms here
- Population frequency
- Population frequency as calculated overall or by population group in the 1000 Genomes and gnomAD databases
- Clinical
- Gene disease panels (genes included in the chosen PanelApp panels)
- Clinical annotation from ClinVar or COSMIC
- Full-text search on HPO, ClinVar, protein domains or keywords, and some OMIM and Orphanet IDs
- Deleteriousness
- Deleteriousness of variant using prediction tools SIFT, Polyphen and CADD (CADD is only available in GRCh37 studies)
- Conservation
- Conservation score as calculated by PhyloP, PhastCons and GERP
Results: retrieving filtered variants¶
Once you have defined your filters, click Search to show the resulting variants in table format.
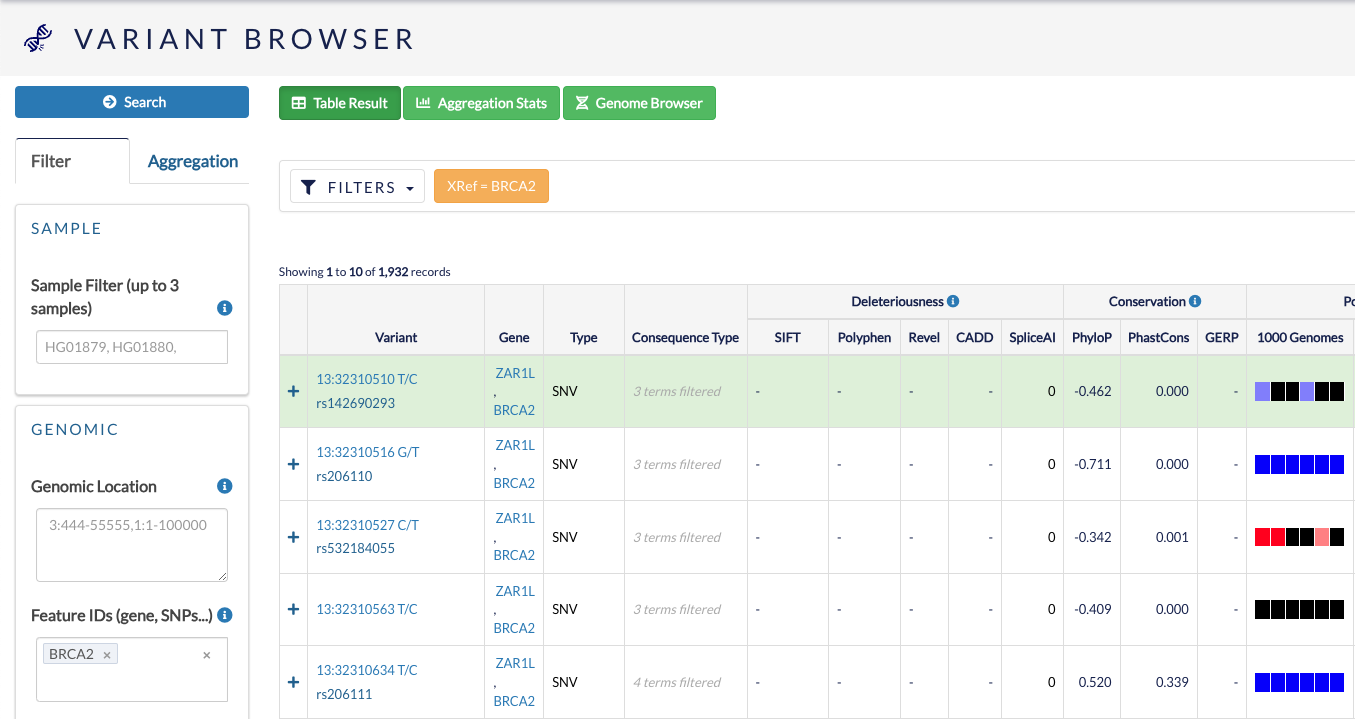
Remember to click Clear if you want to clear your applied filters. If you are experiencing issues with filtering, click Clear and re-try.
You can use this table to browse features of these variants, such as variant type, consequence type and population frequency.
Advanced annotation¶
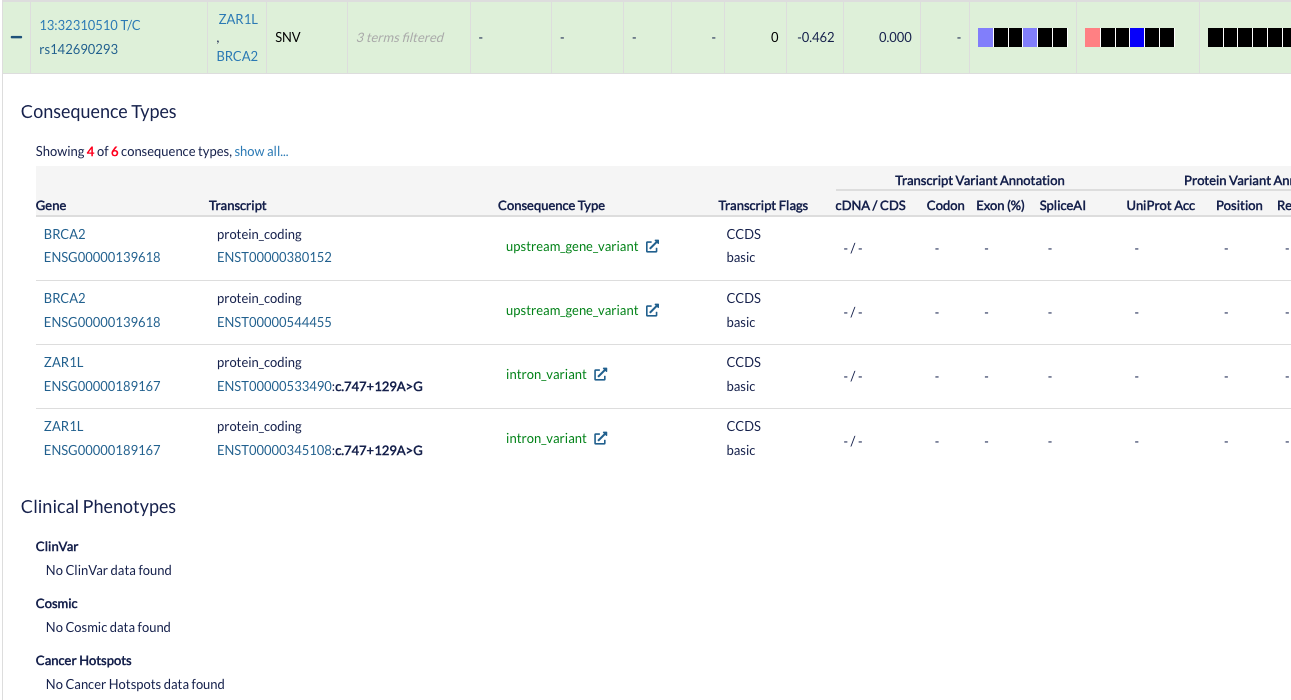
You can see advanced annotation from a variant by pressing the + symbol at the left side of the row corresponding to the variant of interest. You will see a table with the different ENSEMBL transcripts and information about the Clinical Phenotypes associated with that specific variant.
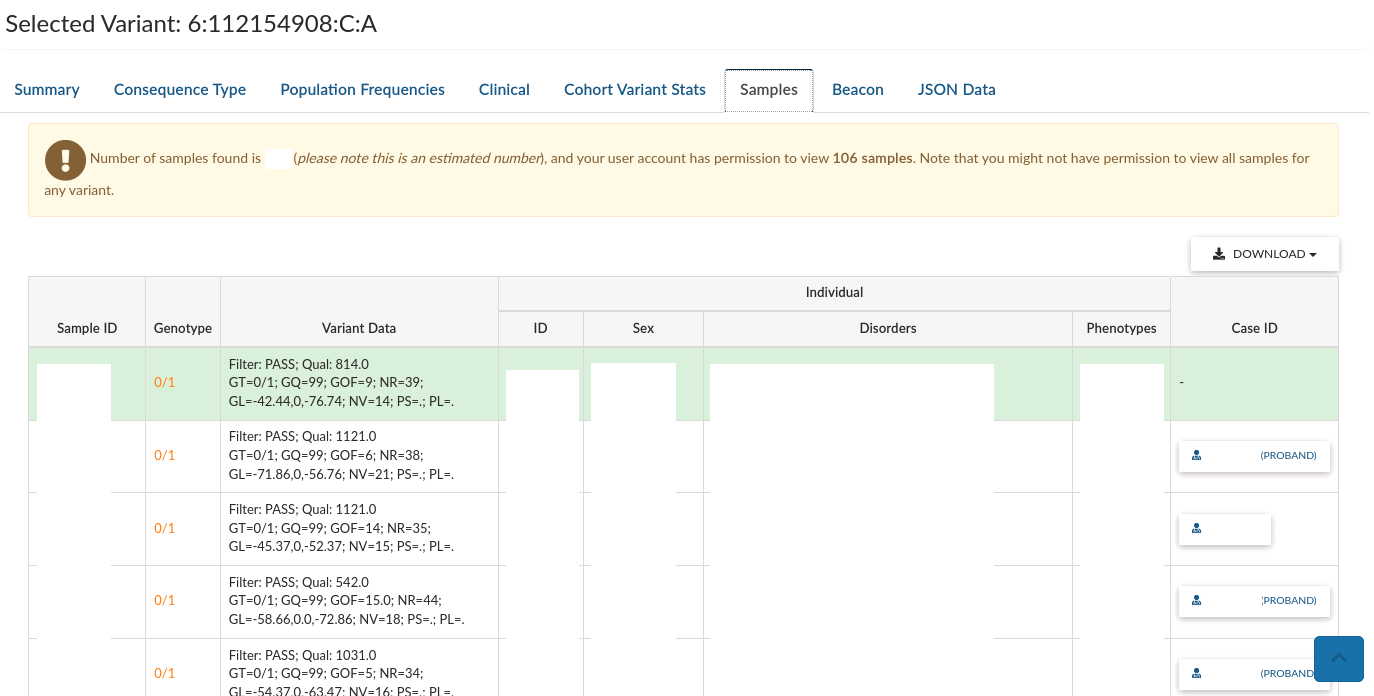
Variant tabs¶
Under the table you will see details of the selected variant. Click on any row of the table to see details for that variant.
-
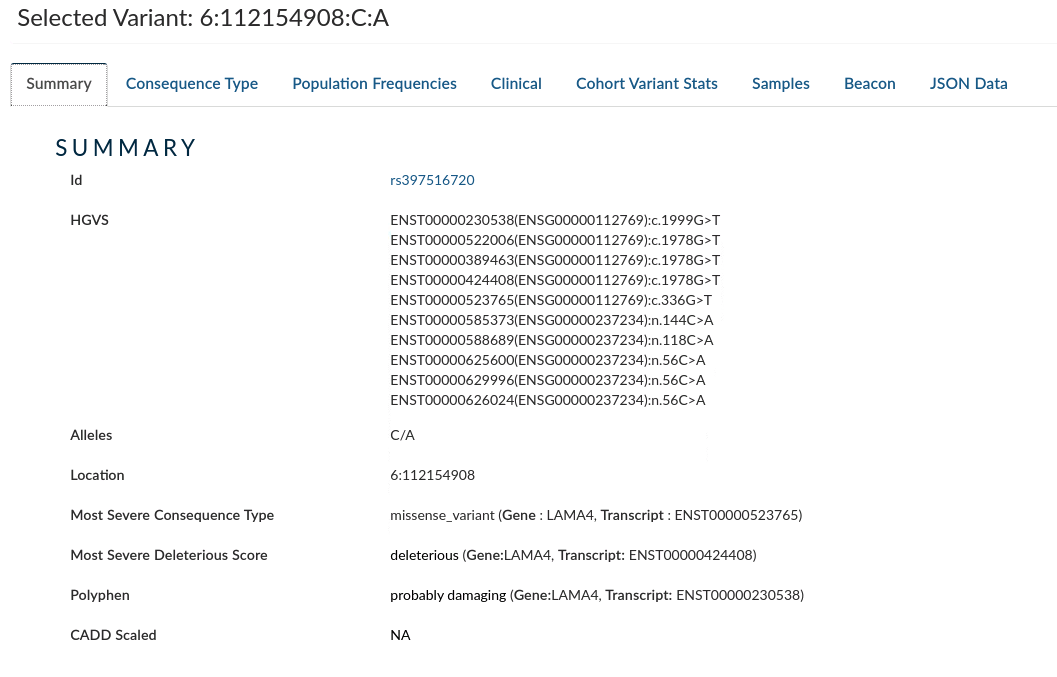
Summary: provides information for the variant from HGVS, Alleles, Location, Most Severe Consequence type, Most Severe deleterious Score, Polyphen and CADD Scaled.
-
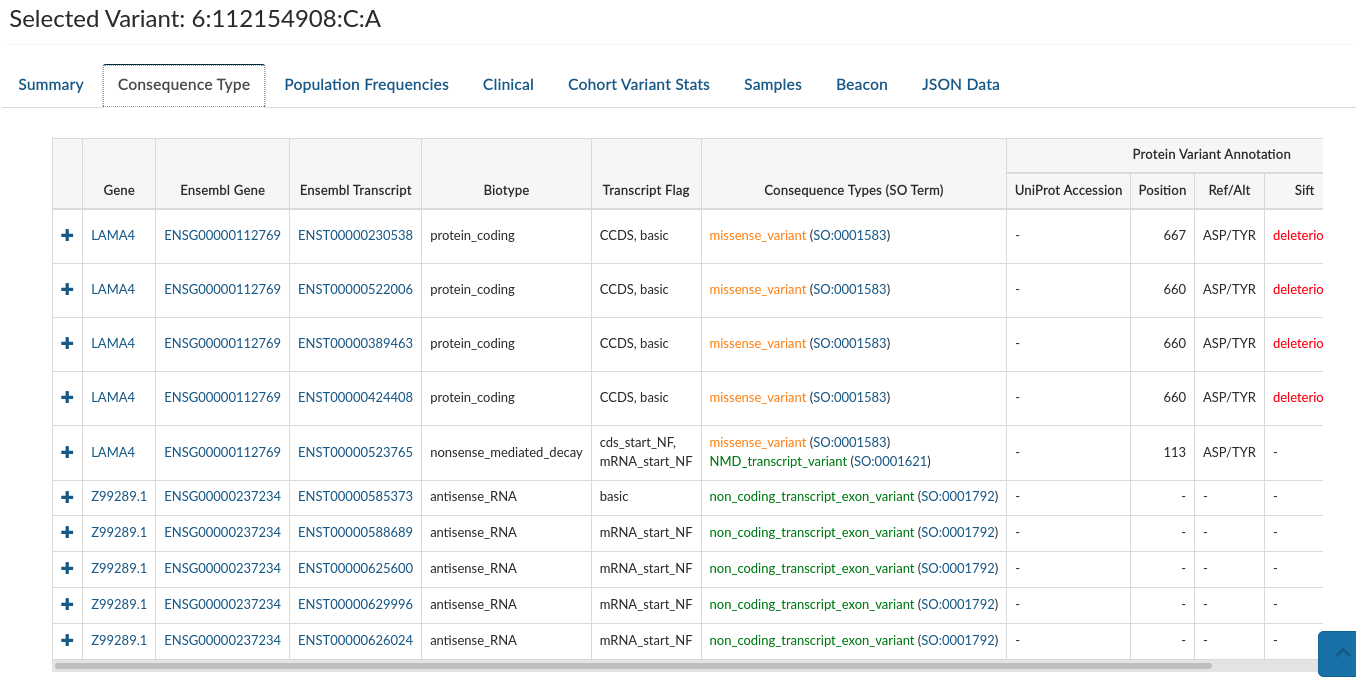
Consequence types: Provides variant annotation at the gene and transcript level.
-
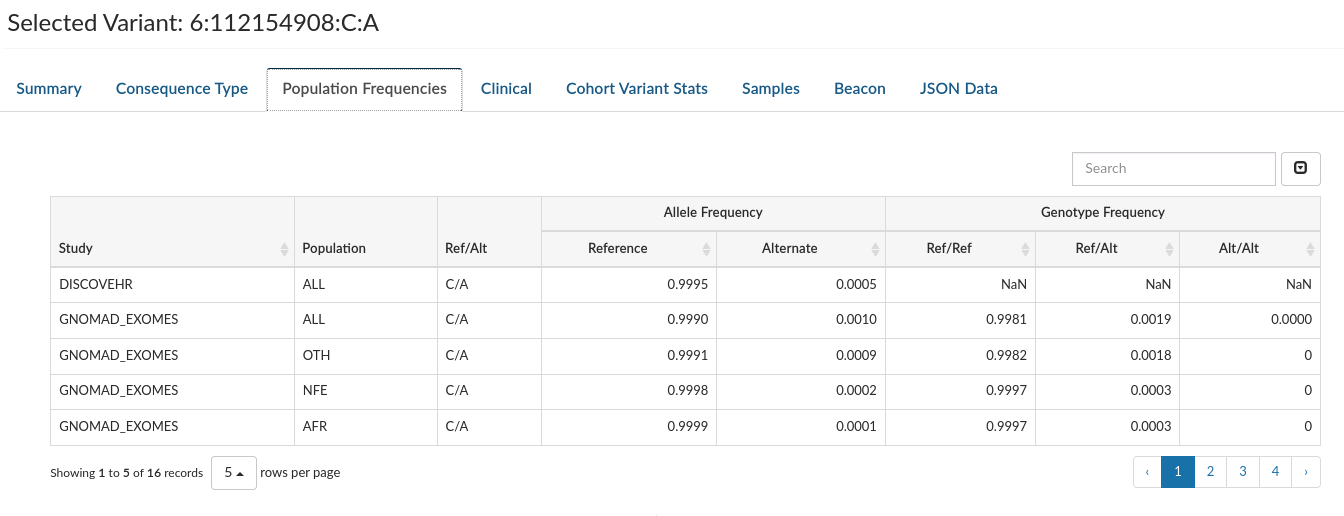
Population Frequencies: Displays frequencies for that variant in all the population studies where it has been observed.
-
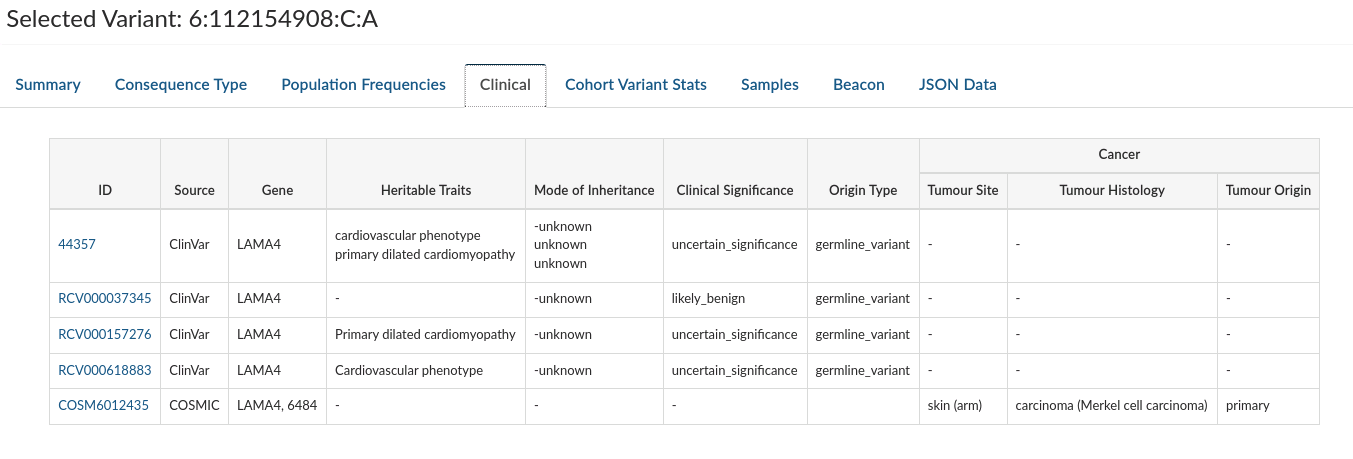
Clinical: Provides information about clinical entries associated with the selected variant in both COMIC and CLINVAR (when they exist).
-
Cohort Variant Stats: Provides the frequency of heterozygous and alternate homozygous across all the samples in the present study, and the other studies from the same project if there exist. It does not take into account reference homozygotes due to the way the data are aggregated, and thus genotype frequencies should be taken with a pinch of salt.
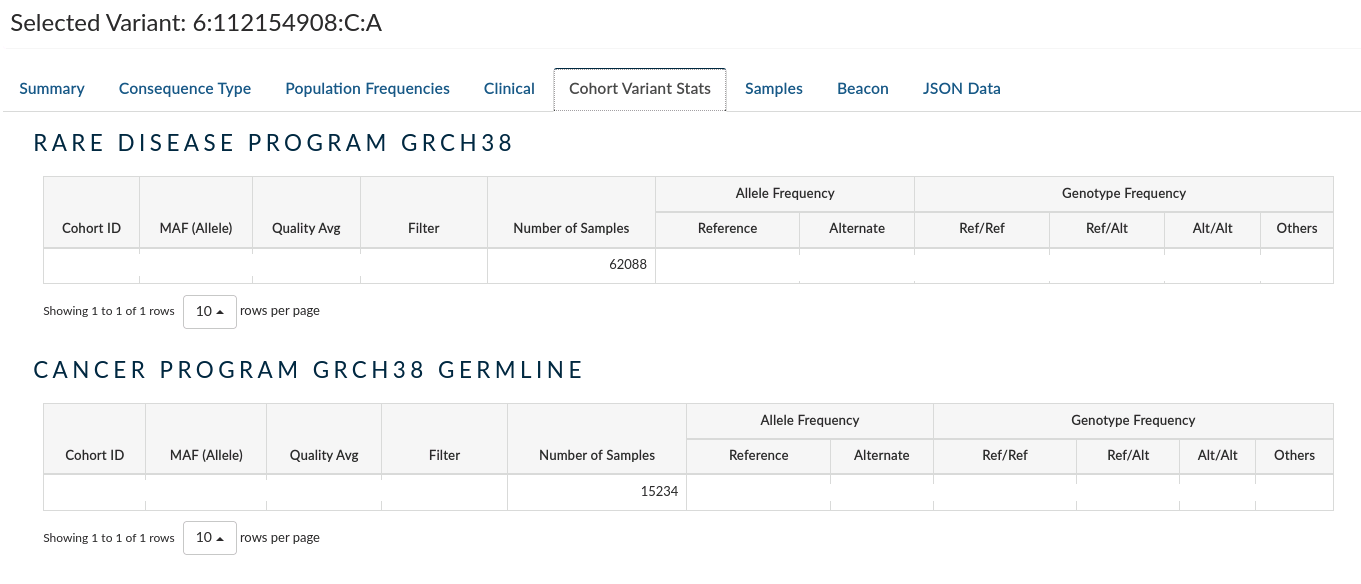
-
Samples gives a list of sample plating IDs that are heterozygous or homozygous alternative for the variant.