AggV2 sample QC¶
We carry out sample-level quality control (QC) on all samples in aggV2. The strategy is based on similar approaches applied on other large scale cohorts (e.g. gnomAD, TOPMed) and on our own observations and investigations on previous aggregations at Genomics England.
Accompanying LabKey table¶
A LabKey table aggregate_gvcf_sample_stats, provided as a companion to aggV2, contains quality statistics for all samples in aggV2. Please see the Data Dictionary for a description of each column.
Sample consistency¶
All 78,195 samples in aggV2 were sequenced with 150bp paired-end reads in a single lane of an Illumina HiSeq X instrument and uniformly processed on the Illumina North Star Version 4 Whole Genome Sequencing Workflow (NSV4, version 2.6.53.23); which comprises the iSAAC Aligner (version 03.16.02.19) and Starling Small Variant Caller (version 2.4.7).
Samples were aligned to the Homo sapiens NCBI GRCh38 assembly with decoys. The contractual obligations from Illumina describe that germline samples must be sequenced to produce at least 85Gb of sequence data with sequencing quality of at least 30. Alignments must cover at least 95% of the genome at 15X or above with well mapped reads (mapping quality >10) after discarding duplicates. Genomes are not delivered to Genomics England if they do not fulfil these obligations.
Additionally, all samples in aggV2 were prepared in the same way.
- DNA was extracted and processed based on the Genomics England Sample Handling Guidelines.
- DNA samples were received in FluidX tubes (Brooks) and accessioned into Laboratory Management Information System (LIMS) at UK Biocentre (Milton Keynes).
- DNA samples undergo quality control assessment (concentration, volume, purity and degradation) at UK Biocentre.
- Upon sample receipt they were visually inspected to ensure the volume was aligned with expectations and a concentration assessment carried out using a dsDNA-specific method.
- DNA samples in the concentration range of 10 ng/ul to 100 ng/ul (Picogreen) were accepted into the sequencing workflows.
- Following automated library preparation, libraries were quantified using automated qPCR, clustered and sequenced. Libraries were prepared using the Illumina TruSeq DNA PCR-Free High Throughput Sample Preparation kit or the Illumina TruSeq Nano High Throughput Sample Preparation kit.
Not all samples are from the same tissue source. Please see below for a detailed breakdown by source.
Participant and genome inclusion criteria¶
- Only 100kGP V10 participants are included
- Participant 'Programme Consent Status' for Research must equal 'Consenting’ from the participant table (this removes samples which are not available to all researchers from the aggregation, such as samples for the Tracer-X participants).
- For known duplicated participants, the participant ID by latest genome delivery by date was used
- The genome platekey ID must exist in the MP V10
plated_samplestable - The genome delivery type / study ID must be in both:
- Bertha Web (Genomics England Internal Bioinformatic Pipeline): rare disease germline | cancer germline
- OpenCGA catalog (Genomics England Internal Bioinformatic Pipeline): RD38 | CG38
- The genome assembly must be:
- Bertha Web (Genomics England Internal Bioinformatic Pipeline): GRCh38 (Illumina NSV4 Pipeline)
- The genome delivery from Illumina must include at least the BAM and the gVCF file
- Genomes with no contamination information are removed
- Genomes with no samtools statistics from Bertha are removed (these have not completed the pipeline fully)
- Genomes with sample type ‘tumour’ are removed
Sample QC filters¶
All 78,195 samples included in aggV2 pass the following quality control filters:
| Sample Attribute | Description |
|---|---|
| Sample Contamination (freemix) | less than 0.03 |
| Ratio of SNV Het to Hom calls | less than 3 |
| Total Number of SNVs | between 3.2M - 4.7M |
| Array Concordance | greater than 90% |
| Median Fragment Size | greater than 250bp |
| Excess of Chimeric Reads | less than 5% |
| Percentage of Mapped Reads | greater than 60% |
| Percentage AT Dropout | less than 10% |
Sample source and library preparation¶
The overwhelming majority of aggV2 are blood samples prepared used EDTA and TruSeq PCR-Free High Throughput (~99% of samples). A small fraction however are prepared from different tissue and library types.
| sample source | sample preparation method | sample library type | number of samples | percent of samples |
|---|---|---|---|---|
| BLOOD | EDTA | TruSeq PCR-Free High Throughput | 77,212 | 98.74 |
| SALIVA | ORAGENE | TruSeq PCR-Free High Throughput | 706 | 0.90 |
| TISSUE | FF | TruSeq PCR-Free High Throughput | 122 | 0.16 |
| FIBROBLAST | FF | TruSeq PCR-Free High Throughput | 72 | 0.09 |
| BLOOD | EDTA | TruSeq Nano High Throughput | 50 | 0.06 |
| GERMLINE | FF | TruSeq PCR-Free High Throughput | 33 | 0.04 |
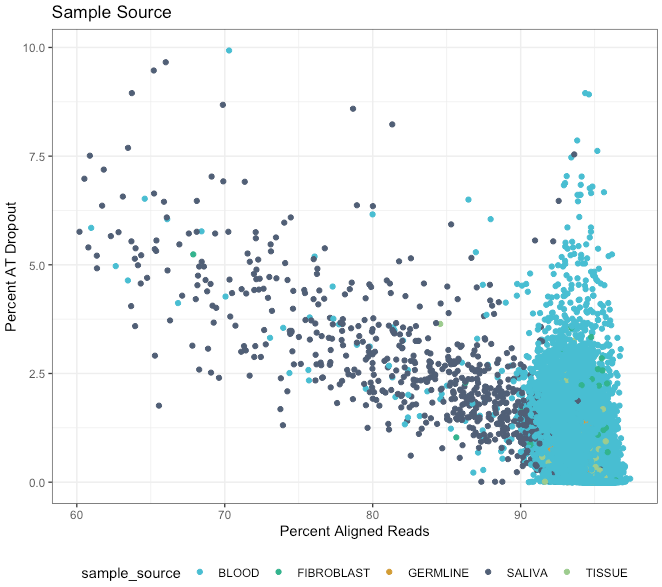
We notice that saliva samples tend to have lower quality than blood ones for some metrics, as shown below for the percent of AT drop out vs percent of aligned reads. Please make sure you take this into account for your analysis where relevant. Use the LabKey table 'aggregate_gvcf_sample_stats' to determine the sample source, preparation method, and library type.
Coverage summary¶
The below graphs and tables describe the coverage summary statistics for the 78,195 samples in aggV2.
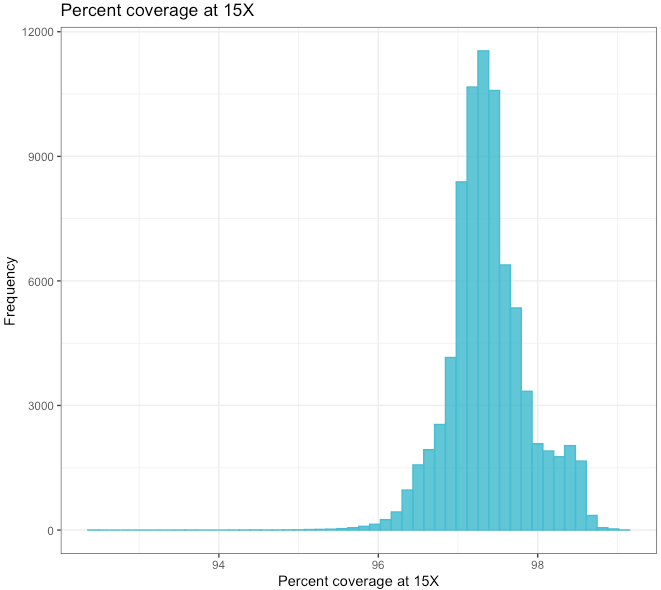
Percent coverage at 15X¶
| Min. | 1st Qu. | Median | Mean | 3rd Qu. | Max. |
|---|---|---|---|---|---|
| 92.48 | 97.10 | 97.34 | 97.38 | 97.63 | 99.14 |
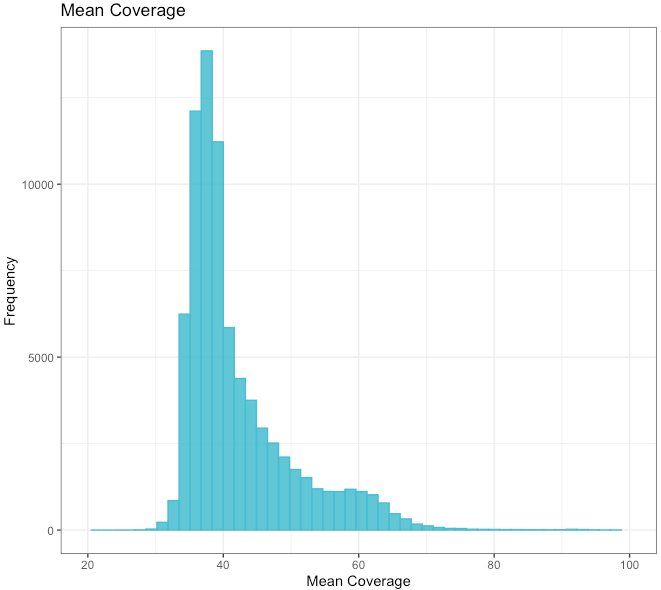
Mean genome-wide coverage¶
| Min. | 1st Qu. | Median | Mean | 3rd Qu. | Max. |
|---|---|---|---|---|---|
| 25.83 | 36.75 | 39.11 | 42.2 | 45.02 | 187.65 |
Help and support¶
Please reach out via the Genomics England Service Desk for any issues related to the aggV2 aggregation or companion datasets, including "aggV2" in the title/description of your inquiry.