COVID-19 aggregations methods¶
Sample information¶
DNA was extracted from whole-blood using EDTA. Libraries were created using the Illumina TruSeq DNA PCR-Free High Throughput Sample Preparation kit and sequenced with 150bp paired-end reads in a single lane of an Illumina HiSeq X instrument (for 100K samples) or NovaSeq instrument (for COVID samples). Alignment and variant calling of small variants was uniformly performed using the Illumina DRAGEN Pipeline (software version 01.011.269.3.2.22 and hardware version 01.011.269). Samples were aligned to the full Homo Sapiens NCBI GRCh38 assembly including decoys, ALT contigs, and EBV sequences.
Participants from the 100K cohort were excluded that had:
- Known cancers of the blood
- Registered a positive COVID-19 test
All samples are derived from blood, using EDTA as the DNA extraction method, and prepared for sequencing using the Illumina TruSeq DNA PCR-Free High Throughput Sample Preparation kit.
Samples just have concordant reported sex (Male/Female) and wgs karyotype sex (XX/XY).
BAM-level quality control¶
Samples were excluded that failed any of the following four BAM-level QC filters in the single-sample:
- Freemix contamination (>3%)
- Mean autosomal coverage (<25X)
- Percent mapped reads (<90%)
- Percent chimeric reads (>5%).
VCF-level quality control¶
A set of VCF-level QC filters were applied post-aggregation on all autosomal bi-allelic SNVs (akin to gnomAD v3.1). Samples were filtered out based on the residuals of eleven QC metrics (calculated by bcftools) after regressing out the effects of sequencing platform and the first three ancestry assignment principle components (including all linear, quadratic, and cross terms) taken from the sample projections onto the 1KG SNP loadings.
Samples were removed that were four median absolute deviations (MADs) above or below the median for the following metrics: ratio heterozygous-homozygous, ratio insertions-deletions, ratio transitions-transversions, total deletions, total insertions, total heterozygous snps, total homozygous snps, total transitions, total transversions. For total singletons (snps), samples were removed that were more than 8 MADs above the median. For ratio of heterozygous to homozygous alternate snps, samples were removed that were more than 4 MADs above the median.
Samples with >5% missingness for SNVs in non-centrometic or telomeric regions were also hard removed.
Aggregation¶
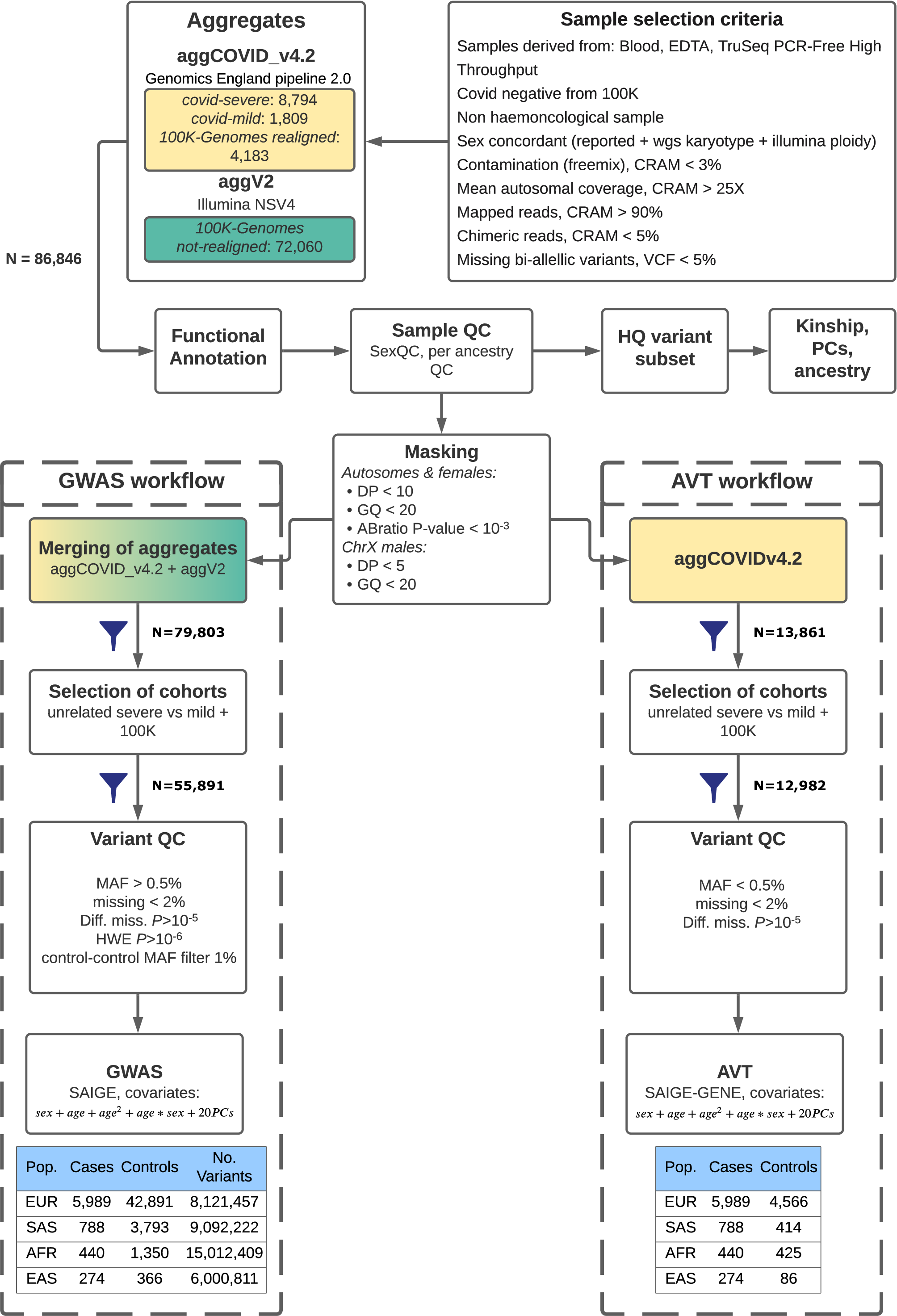
The data provided by Genomics England is organised into genomic aggregates that were generated by joining single sample gVCF files into multi-sample VCF files using GVCFGenotyper (GG) v3.8.1.
The aggregates with prefix aggCOVID_vX contain genomic data (germline WGS data, small variants) for the collection of participants belonging to the COVID-19 severe-cohort, the mild-cohort and cancer participants that were all processed with the Genomics England pipeline 2.0, a custom pipeline for alignment and variant calling using the DRAGEN software (v3.2.22).
As the analysis of the paper required the combined samples from both aggCOVID_v4.2 and aggV2, a joint aggregate was created by merging the respective aggregates using plink1.9 and in all documentation is referred as aggCOVID_v4.2_aggV2.
The diagram below provides a summary of the workflow that generated the data.
Functional annotation¶
In order to maintain parity with the genomic data, we have split the functional annotation data into 1371 chunks. Similarly, the output format of the functional annotation files is compressed vcfs (vcf.gz). The CHROM, POS, REF, ALT, FILTER and INFO fields from the genomic data are preserved for the functional annotation, but genotypes are dropped. All annotations released currently are derived from VEP annotations, as described in the section below. It should be noted that we provide annotations for each variant in the aggCOVID_V4.2 across all transcripts where the variant is found.
Information are written to the INFO/CSQ field, with a '|' field separator.
The specific version of VEP run is: