100kGP Release v10 (03/09/2020)¶
Purpose¶
This document provides a description of the Main Programme Data Release v10.0 dated 3rd September 2020.
Each progressive release incorporates new content, enhances existing content, and enables more effective use of the data.
This data are presented within the Genomics England Research Environment, accessed via the AWS virtual desktop interface and subject to all Genomics England data protection and privacy principles.
Please see the Research Environment User Guide for detailed documentation on how to use and query the Genomics England dataset. This page also includes instructional videos which can not be viewed from within the Research Environment.
Release Overview¶
Data Release Version 10 provides clinical data for 89,256 participants, and 108,431 genomes from 87,383 of these participants. Of these genomes 74,233 are rare disease genomes (from 71,672 participants)1 and 34,198 are cancer genomes (from 15,711 participants)2.
Participants¶
| Type | Count |
|---|---|
| Rare Disease | 71,800 |
| Cancer | 17,339 |
| Total | 89,139 |
Genomes¶
| Type | Genomes count | Participant count |
|---|---|---|
| Cancer Germline | 17,891 | 15,432 |
| Cancer Tumour | 19,333 | 15,593 |
| Cancer Total | 37,224 | 15,612 |
| Rare Disease | 74,008 | 71,419 |
| Genomes Total | 111,232 | 87,031 |
- The genomic data (BAMs, VCFs, and associated quality metrics) delivered to us by our sequencing provider (Illumina) are presented in file shares. These are accessed via your home directory under the subfolder '/genomes/by_date'.
- Clinical data and secondary health data (“medical history”) are presented in LabKey. Tabulated outputs from the Genomics England bioinformatics pipeline are also included in LabKey.
Approximately 10% of the genomic data are aligned against the reference genome version GRCh37 and the remaining majority (90%) against version GRCh38. The alignments were also made using different versions of Illumina’s alignment pipelines V2 and V4, reflecting the versions that were applicable at the time of sequencing. The versions for each genome are identified in the Sequencing Report table. We intend to provide consistently realigned and recalled versions of all our genomes in the future.
Audience¶
The intended audience for this document is researchers that have access to the Genomics England Research Environment.
Identifying this data release¶
The clinical data, secondary data, and tabulated bioinformatic data for this data release, and the paths to the applicable genome files, are found in the following LabKey folder:
main-programme/main-programme_v10_2020-09-03
Subsequent releases will be identified by an incremental increase in the version number and the date of data release.
Relevant genomic data produced by the Genomics England Bioinformatics pipeline (such as rare disease tiering, structural and copy-number variant reports for cancer genomes) are found in your home directory, under the folder 'gel_data_resources' and then 'main_programme' (Genomics England Data).
Frequency of Release¶
Until V6 (Feb 2019), data releases were quarterly. As the data has increased in volume and depth, the time to process and create the data releases has extended. Since V7, there have been releases every 4 months. Now that all consented and eligible 100,000 Genomes Project participants' data are in the research environment, the release schedule is designed to maximise the efficiency with which we add exit questionnaire and new secondary data, as well as introducing new datasets e.g. from the Genomic Medicine Service when it comes.
Samples Removed¶
In Data Release V8, a decision was made to review certain categories of participants and their inclusion in the Genomics England’s main programme data. The following scenarios were reviewed and participants discontinued from release V8 onwards:
- Discontinued samples* (samples which were not determined to be complete enough for continued inclusion in data releases as per the scenarios below)
- For both Cancer and Rare Disease
- Cases with samples that have failed QC with no replacement
- Adults (individuals >=18 at time of release) consented as children
- Cancer only
- Cases for which a “sample not sent” notification has been received
- Rare Disease only
- Cases where the clinical data cannot be verified or resolved to a quality where it is appropriate to include them in the research environment, as determined by the Genomics England clinical team
Data held for these discontinued participants will remain in earlier Main Programme releases but will not be included in this or subsequent data releases.
In addition to the above data withdrawals, in rare occasions, we may have to completely remove the genomes of individuals across all data releases to abide by regulatory rules.
For Data Release v10, we have removed the following two platekey samples from LabKey tables:
LP3000986-DNA_B05 – This concerns a hard removal whereby genome data will be removed in its entirety, due to issues regarding third party consent. The clinical data will remain present and accessible in the system.
LP3000144-DNA_F05 – This concerns a soft removal whereby genome data will remain on the system for analysis done on previous data releases. The variant files for this sample showed truncated data, providing data only up to chromosome 9. Variant data for this sample is likely to return at a later stage in the form of DRAGEN realigned files. The clinical data will remain present and accessible in the system.
*Participants with discontinued samples/data will be informed directly via relevant NHS Genomic Medicine Centres.
Scope¶
In scope¶
Data that are in scope for this release:
- Cancer and rare disease data for the main programme participants with current consent. These data include:
- Genomic data for participants when available
- Whole genome sequencing (WGS) family-based quality control for rare disease, reporting sex checks and pedigree checks
- Outputs of the Genomics England Bioinformatics Research Services
- A new aggregated Illumina gVCF for germline genomes (genomes included are from release 8). Please see the documentation here: Aggregated Variant Calls (aggV2)
- New Principal Components for germline genomes (genomes included are from release 8)
- New ancestry assignments for samples based on genomic data (genomes included are from release 8)
- Genome-wide de novo variant dataset for 13,836 trios from 12,505 families from the rare disease programme. Please see the documentation here: The de novo variant research dataset for the 100,000 Genomes Project
- Outputs of the Genomics England Bioinformatics rare diseases interpretation pipeline
- Tiering data – rare disease
- Exomiser results for interpreted genomes – rare disease
- GMC outcome data ("exit questionnaire data") – rare disease - up until 16/07/2020.
- Outputs of the Genomics England Bioinformatics cancer interpretation pipeline
- Gold standard cancer genomes which have been through interpretation and passed quality checks
- Tumour signature and mutational burden data
- Annotation and tiering of small variants
- Tiering, structural and copy number variant report
- Cancer Principal Component Analysis (PCA). For more information on these metrics please see the following document: Cancer Analysis Technical Information Document.
- Primary clinical data, including formal pedigree data on rare disease participants where it is available; and
- Secondary datasets (medical history), these are available at varying levels of completeness and include:
- Hospital Episode Statistics (HES), including HES Accident and Emergency, HES Admitted Patient Care, and HES Outpatient Care.
- Diagnostic Imaging Dataset (DID).
- Patient Reported Outcome Measures (PROMs).
- Mental Health Minimum Dataset (MHMDS).
- Mental Health Learning Disabilities Dataset (MHLDDS).
- Office for National Statistics - Death details data (ONS).
- Systemic Anti-Cancer Therapy Dataset (SACT).
- Systemic Anti-Cancer Therapy Dataset - UNCURATED (SACT_UNCURATED).
- National Radiotherapy Dataset (RTDS).
- Cancer Registration (AV) tables.
- Cancer waiting times (CWT).
- Lung Cancer Data Audit (LUCADA).
- National Cancer Registration and Analysis Service Diagnostic Imaging Dataset (NCRAS_DID).
- Sample datasets describing:
- Handling and quality control of DNA samples at the Genomic Medicine Centres, the biorepository and the sequencer.
- Omics samples stored at the biorepository.
- Orthogonal standard-of-care test data collected from GMCs for a subset of cancer patients
Out of scope¶
Additional time is required to update the applications/tools that are available in the RE to the current data release, e.g. IVA, Participant Explorer. Please refer to the Application Data Versions page for the data release version used in the RE products and services.
Data out of scope for this release:
- Clinical and genomic data for participants that have withdrawn from the 100,000 Genomes Project or were otherwise ineligible (n=8351).
- Participant data from the pilot phases of the project (i.e. not main programme, n=527).
Quality Notes¶
- BAM and VCF genomic data files are as they have been delivered to us by our sequencing provider (Illumina). These have all passed an initial QC check based on sequencing quality and coverage. They have, however, not all undergone our full in-house quality checks and they are therefore subject to potential discrepancies or inaccuracies. Such checks include, but are not limited to, discrepancies in genetic versus reported sex and in family relationships.
- As participants undergo the in-house checks and pass through the Genomics England interpretation pipeline, any inaccuracies we identify will be rectified in subsequent releases.
- Any samples that have been affected prior to this release (e.g. sample swaps or samples that have been retracted as part of the in-house QC process) are listed in Section 10 below.
- You are encouraged to work on the subset of samples that have already passed our internal QC checks; these can be found below for rare disease and cancer genomes, respectively.
- For Rare Disease genomes, you should note that all tiered genomes have passed through Genomics England in-house QCs and that all tiered genomes come from the pool of genomes that have had family checks applied to them, as a first step towards Genomics England tiering. For rare disease interpretation including tiering, small variants are called using the Platypus variant caller. Please see the Rare Disease Results Guide on our Further reading and documentation page for more information.
- Different QC filtering has been applied to the Illumina VCF files and the Platypus VCFs that are used for tiering. There may therefore, be tiered variants that have been filtered out of the Illumina VCF files, and, conversely, variants present in the Illumina VCF file that have been filtered out of the platypus VCFs.
- Some rare disease families lack a proband due to the availability of data at the time of release. The missing data will be made complete in a future release if available.
- Human Phenotype Ontology (HPO) terms may be missing or incomplete for some participants.
- Pedigree data are only available for a subset of rare disease participants. Each participant’s relationship to their family’s proband is available for such cases in the rare_diseases_pedigree_member table; this can be used to determine family relationships instead of formal pedigree data.
- WGS family selection quality checks are provided for rare disease genomes on GRCh38, reporting abnormalities of sex chromosomes and reported vs genetic sex summary checks (computed from family relatedness, Mendelian inconsistencies, and sex chromosome checks). Full details on why a family has failed a reported vs genetic sex check can be requested via the Service Desk.
- For Cancer genomes, you should note that all 'gold standard genomes' that have been through Genomics England interpretation and passed quality checks are found in the cancer quick view table cancer_analysis. We strongly recommend using the data from this table for all cancer analyses.
- Clinical data and secondary data have been provided as submitted and have undergone limited validation and cleaning
- sact_uncurated is the table with the raw feed from NCRAS which feeds into their curation process producing the SACT table (both under PHE/NCRAS section), which remains the gold standard. A major point to raise is that this SACT curation does not provide tumour IDs, thus you must match this dataset to other NCRAS registries by adjusting for date. A lot of familiar data fields remain in their raw non-standardised form (sex, treatmentintent, clinicaltrialindicator). Pending feedback, these fields can be normalised in subsequent updates.
Conditions of Use¶
Participants identified as TracerX in the field normalised_consent_form in the participant table in LabKey must not be used by commercial organisations. Commercial organisations do not have access to the genomic data of TracerX participants.
Participants with a participant ID that commences with 125 or 226 were recruited through the Scottish Genomes Partnership Research Programme. These are under the governance of a separate but linked consent and protocol to the 100,000 genomes project. Only the removal of summary level statistics is permitted. Airlock approval will not be granted for the removal of record level data associated with these participants.
Data Release Description¶
The Genomics England data are organised into data views (displayed within LabKey as tables) categorised into Quick View, Common, Bioinformatics, Rare Disease and Cancer. The Data Dictionary that describes the table structure and provides data definitions for this release can be found here.
Quick View¶
Data views that bring together data from several LabKey tables for convenient access:
| Name of Table / Data View | Description |
|---|---|
| rare_disease_analysis | Data for all rare disease participants including: sex, ethnicity, disease recruited for and relationship to proband; latest genome build, QC status of latest genome, path to latest genomes and whether tiering data are available; as well as family selection quality checks for rare disease genomes on GRCh38, reporting abnormalities of the sex chromosomes, family relatedness, Mendelian inconsistencies and reported vs genetic sex summary checks. Please note that only sex checks are unpacked into individual data fields; a final status is shown in the “genetic vs reported results” column. |
| cancer_analysis | Data for all cancer participants whose genomes have been through Genomics England bioinformatics interpretation and passed quality checks, including: sex, ethnicity, disease recruited for and diagnosis; tumour ID, build of latest genome, QC status of latest genome and path to latest genomes; as well file paths to the genomes. This table includes information derived from laboratory_sample and cancer_participant_tumour. Some key data included in the table are elucidated below: Global Tumour Mutation Burden This is the number of somatic non-synonymous small variants per megabase of coding sequences (32.61 Mb). This metric was calculated using somatic_small_variants_annotation_vcf as input (see below for description) and all non-PASS variants were removed from the calculation. Tumour purity This is the tumour purity (cancer cell fraction) as calculated by Ccube Mutational Signatures The table includes the relative proportions of the different mutational signatures demonstrated by the tumour. Analysis of large sequencing datasets (10,952 exomes and 1,048 whole-genomes from 40 distinct tumour types) has allowed patterns of relative contextual frequencies of different SNVs to be grouped into specific mutational signatures. Using mathematical methods (decomposition by non-negative least squares) the contribution of each of these signatures to the overall mutation burden observed in a tumour can be derived. Further details of the 30 different mutational signatures used for this analysis, their prevalence in different tumour types and proposed aetiology can be found at the Sanger Institute Website. Cancer PCA QC Statistics The cancer analysis pipeline employs a sequencing quality control check which selects several important statistics associated with the sequencing returned by the sequencing provider, and uses them to check whether or not the sample in question is an outlier with respect to previous samples that have been run through the pipeline. It is, in effect, a safety net that can spot issues that have occurred at the tissue collection stage (i.e. at the GMC (Genomic Medicine Centre)) or at the library preparation step (i.e. at the sequencing provider), both of which may impact upon the final genomic analysis returned to the clinician. Somatic small variants annotation vcf filepaths The somatic_small_variants_annotation_vcf column contains file paths pointing to VCFs containing Genomics England flags for potential false positive variants as well as additional annotations (see VCF header for details). SIFT and PolyPhen scores as well as new PONnoise50SNV flag were added. The flags used for annotation are: i. CommonGermlineVariant: variants with a population germline allele frequency above 1% in an early subset of the Genomics England dataset. ii. CommonGnomADVariant: variants with a population germline allele frequency above 1% in gnomAD dataset iii. RecurrentSomaticVariant: recurrent somatic variants with frequency above 5% in an early subset of the Genomics England dataset iv. SimpleRepeat: variants overlapping simple repeats as defined by Tandem Repeats Finder v. BCNoiseIndel: small indels in regions with high levels of sequencing noise where at least 10% of the basecalls in a window extending 50 bases to either side of the indel’s call have been filtered out by Strelka due to the poor quality vi. PONnoise50SNV: SNVs resulting from systematic mapping and calling artefacts The following methodology was used for the PONnoise50SNV flag: the ratio of tumour allele depths at each somatic SNV site was tested to see if it is significantly different to the ratio of allele depths at this site in a panel of normals (PoN) using Fisher’s exact test. The PoN was composed of a cohort of 7000 non-tumour genomes from the Genomics England dataset, and at each genomic site only individuals not carrying the relevant alternate allele were included in the count of allele depths. The mpileup function in bcftools v1.9 was used to count allele depths in the PoN, and to replicate Strelka filters duplicate reads were removed and quality thresholds set at mapping quality >= 5 and base quality >= 5. All somatic SNVs with a Fisher’s exact test phred score < 50 were filtered, this threshold minimised the loss of true positive variants while still gaining significant improvement in specificity of SNV calling as calculated from a TRACERx truth set. A presentation entitled PONnoise50SNV: SNVs resulting from systematic mapping and calling artefacts, which further outlines the methodology, can be found in the Publications and other useful links table located on our Further reading and documentation page. Alignment BAM files generated by Isaac Genome Alignment Software A paper written by GECIP members discussing the issue of reference bias in the computation of variant allele frequencies (VAFs) by the Illumina Isaac pipeline (caused by preferential soft clipping of reads supporting alternate alleles) can be located here |
Common¶
Data views that are common to both the rare disease and the cancer domains. This data pertains to sample handling, genome sequencing, and participant data.
Data Relating to Participants:
| Name of Table / Data View | Description |
|---|---|
| participant | Data on each individual participant in the 100,000 Genomes Project, e.g. personal information (such as relatives or self-reported ethnicity); points of contact with the Project (e.g. handling Genomic Medicine Centre or Trust); and a record of the status of their clinical review. |
| death_details | Data on participant deaths submitted by GMCs, likely less complete than the data collected by ONS and NHSE. |
Data Relating to Samples:
| Name of Table / Data View | Description |
|---|---|
| clinic_sample | Data describing the taking and handling of participant samples at the Genomic Medicine Centres, i.e. in the clinic, as well as the type of samples obtained. Because of the complexities of handling and managing tumour tissues samples in a clinical setting, there are many fields that are cancer-specific. |
| clinic_sample_quality_check_result | Data describing the quality control of obtaining and handling participant samples at the Genomic Medicine Centres, i.e. in the clinic. |
| laboratory_sample | Data describing the handling of samples at the biorepository and in preparation for sequencing, as well as the type of sample. |
| plated_sample | Data describing the handling and QC of samples at Illumina (the sequencing provider). |
| laboratory_sample_omics_availability | Availability of samples collected from participants in the 100,000 Genomes Project for the purpose of omics research. Data includes: Participant ID, Sample Type (e.g. Serum, RNA Blood), the number of aliquots of that sample type for that participant, and the availability status - whether the sample has already been used for a research project. Research proposals for the use of these samples can be submitted, via the GECIP team, to the Scientific Advisory Committee and Access Review Committee. |
| lrs_laboratory_sample | Data describing the characteristics and processing methods (DNA to library preparation) of samples from participants in the 100,000 Genomes Project for which long-reads sequencing has been carried out. |
Bioinformatics¶
Contains tables with data that are related to the genomic data and the outputs from the Genomics England interpretation pipeline data for participants from both cancer and rare disease programmes. These tables do not directly include primary and secondary sources of clinical data.
| Name of Table / Data View | Description |
|---|---|
| sequencing_report | For each participant in the 100,000 Genomes Project, this table contains data describing the sequencing of their genome(s) and associated output, as well as the sample type that the sequence is from. |
| genome_file_paths_and_types | Data that specifies the genomic files and their folder locations for a given participant. Please be aware that the same genome can be released with multiple versions of mapping/variant calling pipeline. Since the Main Programme Data Release Version 10, we have added file paths to genomes that have been realigned with the DRAGEN pipeline. Please see the change summary for Main Programme Data Release v10 below on how to select for these. |
| aggregate_gvcf_sample_stats | This table accompanies the aggregated Illumina gVCFs (/gel_data_resources/main_programme/aggregation/aggregate_gVCF_strelka/aggV2). Individual sample QC data was retrieved from Genomics England OpenCGA database. Most sequencing metrics are BAM file statistics provided from Illumina or Genomics England WGS data processing pipeline. The table contains principal components, a set of unrelated individuals and probabilities of ancestry membership, and more. (These are crude categories to represent broad groups of ancestries. Please do not over-interpret these.) Please also refer to the documentation listed here: Aggregated Variant Calls (aggV2). This table was changed significantly from Main Programme Data Release Version 10 onwards. Please see the change summary of the respective Data Release below. |
| tiered_data | For each participant of the 100,000 Genomes Project who has been through the Genomics England interpretation pipeline, this table contains data describing the variants that are identified as plausibly pathogenic for a participant's phenotype. The tiering process is based on a number of features such as their segregation in the family, frequency in control populations, effect on protein coding, and mode of inheritance. and whether they are in a gene in the virtual gene panel(s) applied to the family. The applied panels can be found in the respective table 'panels_applied'. |
| tiered_variants_frequency | This table contains the frequencies of each tiered variant for every Project participant for whom we provide tiered variants. |
| panels_applied | For each participant of the 100,000 Genomes Project, this table contains the name and version of the panel(s) that was applied to his or her genome. |
| exomiser | This table contains the full results from the Exomiser rare disease SNV and Indel Prioritisation Process. All rare disease cases are now run through the Exomiser automated variant prioritisation framework developed by members of the Monarch initiative: principally Dr. Damian Smedley’s team at Queen Mary University London and Professor Peter Robinson’s team at Jackson Laboratory, USA, with previous contributions from staff at Charité –Universitätsmedizin, Berlin and the Sanger Institute. Given a multi-sample VCF file, family pedigree and proband phenotypes encoded by Human Phenotype Ontology (HPO) terms, Exomiser annotates the consequence of variants (based on Ensembl transcripts) and then filters and prioritises them for how likely they are to be causative of the proband’s disease based on: 1) the predicted pathogenicity and allele frequency of the variant in reference databases 2) how closely the patient’s phenotypes match the known phenotypes of diseases and model organisms associated with the gene. Please see Publication Website |
| gmc_exit_questionnaire | Data reporting back from the Genomic Medicine Centres, for variants reported to them by Genomics England, to what extent a family’s presenting case can be explained by the combined variants reported to them (including any segregation testing performed); confidence in the identification and pathogenicity of each variant; and the clinical validity of each variant or variant pair in general and clinical utility in a specific case (only the most recent update will be shown and only one questionnaire per report). |
| domain_assignment | For each participant in the 100,000 Genomes Project, this table contains: data describing the disease type to which they were recruited; the gene panel(s) applied to their genome(s); the GECIP domain to which their genome(s) have been assigned for the purposes of administering the GECIP publication moratorium; whether this participant is still under moratorium as of the date of release, and the end date of the GECIP moratorium associated with their genome(s). |
| cancer_staging_consolidated | This table combines staging information from our primary clinical data (cancer_participant_tumour) and secondary clinical data from NCRAS (sact and av_tumour) to give a stage for each sample we have sequenced and fully interpreted on our database (cancer_analysis). The staging information may be in form of TNM combined, each component or other standards such as AJCC, or Dukes', for example. The genomic data is matched to the clinical data using a disease type (genomic data) and ICD code (clinical data) correspondence dictionary created and validated internally. Also, the clinical stage information must not be further away than one year from the date the sample has been collected. Note that, the column names have been preserved as found in the original datasets they were extracted from, except for tumour_pseudo_id found both in sact and av_tumour, where a prefix with the dataset names was added to. Also, for each staging dataset used, when more than one entry for the same patient was available the closest one to the clinical data collection has been kept. Further information on the staging table and its generation process can be found in the document: Staging data (Cancer) |
| denovo_cohort_information | Table with cohort information for all participants included in the de novo variant dataset. Attributes within this table include: participant ID, sex, affection status, family ID, pedigree ID, and the path to each family's multi-sample VCF with flagged DNVs. See De novo variant research dataset for more information. |
| denovo_flagged_variants | Table of all base_filter pass variants for all trios within the DNV dataset. This table includes all flags from the DNV annotation pipeline for each variant. See De novo variant research dataset for more information. |
| lrs_sequencing_data | This table includes data describing long-read sequencing of a subset of 100,000 Genomes Project participants and associated output, including paths to raw and BAM files. |
Rare Diseases¶
Rare Disease data are presented at the level of Rare Disease families (families of probands), Rare Disease pedigrees, and participants. Participants are individuals who have consented to be part of the project with the expectation that a sample of their DNA will be obtained and their genome sequenced. Pedigree members are extended members of the proband’s family, this includes participants as well a small amounts of deidentified data recorded to allow a full picture of the proband’s extended family. This additional information is extracted from the proband’s medical record.
All Rare Disease table names are prefixed with “rare_diseases_”.
Data at the Level of Rare Disease Families:
| Name of Table / Data View | Description |
|---|---|
| rare_diseases_family | Data describing the families of rare disease probands participating in the 100,000 Genomes Project. It includes the family group type, the status of the family’s pre-interpretation clinical review and the settings that were chosen for the interpretation pipeline at the clinical review. |
| rare_diseases_pedigree | Data describing the Rare Disease participants, linking pedigrees to probands and their family members. |
| rare_diseases_pedigree_member | Data describing the Rare Disease pedigree members, similar to the data about each individual participant in the participant table (common data view, see section 8.2). It may also include additional data, such as the age of onset of predominant clinical features; data on links to other family members; as well as data collected only for Phenotypes. |
Data at the Level of Rare Disease Participants.
The data presented in these tables provides information on disease progression and pertinent medical history:
| Name of Table / Data View | Description |
|---|---|
| rare_diseases_participant_disease | Data describing the rare disease participants' disease type/subtype assigned to them upon enrolment, and the date of diagnosis. |
| rare_diseases_participant_phenotype | Data describing the Rare Disease participants’ phenotypes. For each Rare Disease participant in the 100,000 Genomes Project, there are data about whether a phenotypic abnormality as defined by an HPO term is present and what the HPO term is, as well as the age of onset, the severity of manifestation, the spatial pattern in the body and whether it is progressive or not. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_gen_measurement | For Rare Disease participants in the 100,000 Genomes Project, this table contains general measurements relevant to the disease, alongside the date that the measurements were taken on. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_early_childhood_observation | For Rare Disease participants in the 100,000 Genomes Project, this table contains measurements and milestones provided by the GMCs, related to childhood development. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_imaging | For Rare Disease participants in the 100,000 Genomes Project, this table contains various data and measurements from past scans, alongside the date of the scans. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_invest_genetic | For Rare Disease participants in the 100,000 Genomes Project, this table contains information on any genetic tests carried out. Data characterising the genetic investigation is recorded alongside records of the sample tissue source and the type of testing laboratory. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_invest_genetic_test_result | For Rare Disease participants in the 100,000 Genomes Project, this table contains the results of any genetic tests carried out. Following on from the rare_diseases_invest_genetic table, a summary of the results is presented and contextualised by testing method and scope. Please note that these data are only available for a subset of the rare disease participants. |
| rare_diseases_invest_blood_laboratory_test_report | For Rare Disease participants in the 100,000 Genomes Project, this table contains the results of any blood tests carried out. Over 400 blood values are recorded alongside type and technique of testing and the status of the participating patient in the care pathway. Please note that these data are only available for a subset of the rare disease participants. |
Cancer¶
Cancer data are presented for either the patient level cancer diagnosis/disease type or the tumour-specific sample details of participants in the Cancer arm of the 100,000 Genomes Project.
All Cancer table names are prefixed with “cancer_"
Data Relating to Cancer Participants:
| Name of Table / Data View | Description |
|---|---|
| cancer_participant_disease | For each cancer participant in the 100,000 Genomes Project, this table includes data about their cancer disease type and subtype. |
| cancer_participant_tumour | For each cancer participant’s tumour in the 100,000 Genomes Project, this table contains data that characterises the tumour, e.g. staging and grading; morphology and location; recurrence at time of enrolment; and the basis of diagnosis. |
| cancer_participant_tumour_metastatic_site | For each cancer participant in the 100,000 Genomes Project, this table contains the site of their metastatic disease in the body (if applicable) at diagnosis. |
| cancer_care_plan | For a proportion of cancer participants in the 100,000 Genomes Project, this table contains information from their NHS cancer care plan on their treatment and care intent, in particular outcomes of MDT meetings and coded connected data (e.g. diagnoses from scans). |
| cancer_surgery | For a proportion of cancer participants in the 100,000 Genomes Project, this table contains details of what surgical procedures were had, as well as the specific location of the intervention. |
| cancer_risk_factor_general | For a proportion of cancer participants in the 100,000 Genomes Project, this table contains data on general cancer risk factors, namely smoking status, height, weight and alcohol consumption. This table was compiled with input from GECIP members. |
| cancer_risk_factor_cancer_specific | For a proportion of cancer participants in the 100,000 Genomes Project, this table contains data on specific risk factors related to particular cancer types. This table was compiled with input from GECIP members. |
| cancer_invest_imaging | For a proportion of cancer participants in the 100,000 Genomes Project, this table contains: coded data on imaging investigations characterising the scan, its modality, anatomical site and outcome; as well as the outcome of the imaging report in free text form. |
Data derived from or relating to tumour samples:
| Name of Table / Data View | Description |
|---|---|
| cancer_invest_sample_pathology | For a subset of cancer participants in the 100,000 Genomes Project, this table contains full pathology reports and other related data on and from their tumour samples around diagnosis and characterisation of the cancer. Please note that much of this information is also found in the clinic_sample and cancer_participant_tumour tables. |
| cancer_specific_pathology | For a subset of tumours from cancer participants in the 100,000 Genomes Project, this table contains pathology data specific to that participant’s cancer type. This may provide additional data to the cancer_invest_sample_pathology and cancer_participant_tumour tables. |
| cancer_systemic_anti_cancer_therapy | For a subset of tumours from cancer participants in the 100,000 Genomes Project, this table contains details the regimen and intent of the patients’ chemotherapy. |
| cancer_invest_circulating_tumour_marker | For a subset of tumours from cancer participants in the 100,000 Genomes Project, this table contains biomarker measurements specific to particular cancer types. |
Secondary Data¶
Secondary data tables are the corpus of curated data we receive from national data warehouses for all eligible participants not belonging in a data restricting cohort and not registered in Northern Ireland, Wales or Scotland. They are mostly longitudinal in nature and agnostic to the recruited disease. Data at the point of release captures all activity contained in the period covered within each of the datasets up to the latest quarter published by NHSE and end of calendar year for PHE/NCRAS.
NHSE¶
- HES: Hospital Episode Statistics containing details of all commissioned activity during admissions, outpatient appointments and A&E attendances.
- DID: Metadata (demographics, modalities, ordering entity and dates) on diagnostic imaging tests collated from local radiology information systems.
- PROMS: Patient Reported Outcome Measures report health gain in patients undergoing major surgical operations based on responses to questionnaire pre and post procedure.
- MHMDS: Data on patients receiving care in NHS specialist mental health services. Reporting care period for this dataset is up to March '14.
- MHLDDS: Data on patients receiving care in NHS specialist mental health services. Reporting care period for this dataset is from March '14 to Dec '15'. Will be replaced in the future with MHSDS.
- ONS/CEN: Office of National Statistics registry data for cancer registrations and deaths inside and outside hospitals. Issue of death certificates and cancer network registrations are a requirement for an entry to these manifests.
| Name of Table / Data View | Description |
|---|---|
| hes_apc | Historic records of admissions into secondary care of GEL main programme participants. |
| hes_cc | Historic records of admissions into critical care of GEL main programme participants. |
| hes_op | Historic records of outpatient attendances of GEL main programme participants. |
| hes_ae | Historic records of A&E attendances of GEL main programme participants. |
| did | Historic diagnostic Imaging records of GEL main program participants. |
| did_bridge | Linking file of participants to DID submissions. |
| proms | Questionnaire responses pre and post four operations: hip replacement, knee replacement, varicose vein and groin hernia surgery. |
| mhmd_v4_record | Historic records of MH related admissions of GEL main programme participants. One record per spell per patient in a provider. |
| mhmd_v4_event | Historic records of MH related admissions of GEL main programme participants. Episode and event tables link to the records table via spell_id. |
| mhmd_v4_episode | Historic records of MH related admissions of GEL main programme participants. Episode and event tables link to the records table via spell_id. |
| mhldds_record | Historic records of MH related admissions of GEL main programme participants. One record per spell per patient in a provider. |
| mhldds_event | Historic records of MH related admissions of GEL main programme participants. Episode and event table link to the records table via mhm_mhmds_spell_id. |
| mhldds_episode | Historic records of MH related admissions of GEL main programme participants. Episode and event tables link to the records table via mhm_mhmds_spell_id. |
| mh_bridge | Linking file of participants to MHMD records and the three interlinking tables (spells). |
| cen | Cohort Event Notification for GEL main programme participants. Captured events are death and cancer registrations. Death events are associated with date and references to the death register district and number are included. Cancer events are associated with date of cancer registration, reference number and basic cancer type characteristics - site, morphology and behaviour. |
| ons | Office of National Statistics - death registration and cause of death reports for the GEL main programme participants. This table has been truncated to contain date of death and cause of death details only. |
NHSE/NCRAS¶
Available for patients diagnosed with Cancer (ICD10 C00-97, D00-48) from 1 January 1995 - 31 December 2017.
This dataset brings together data from more than 500 local and regional datasets to build a picture of an individual’s treatment from diagnosis.
Please note that pseudo_tumour_ids in AV tables and in SACT are assigned to participants by NCRAS and do not link to the tumour_ids assigned by GEL for sequencing and clinical data. Whilst (particularly in the case of single tumour) this may refer to the same cancer, caution should be applied prior to any analysis.
| Name of Table / Data View | Description |
|---|---|
| av_patient | Patient information - demographics and death details. |
| av_tumour | Tumour catalogue and characterisation for all patients with registerable tumour. Table's |
| av_treatment | Tumour linked catalogue of treatments and sites that provided them for all patients with registerable tumour. |
| av_imd | The Income Deprivation Domain (IMD table) measures the proportion of the population experiencing deprivation relating to low income. The definition of low income used includes both those people that are out-of-work and those that are in work but who have low earnings. |
| av_rtd | Routes to Diagnosis: cancer registration data are combined with Administrative Hospital Episode Statistics data, Cancer Waiting Times data and data from the cancer screening programmes. Using these datasets cancers registered in England which were diagnosed in 2006 to 2016 are categorised into one of eight Routes to Diagnosis. The methodology is described in detail in the British Journal of Cancer article 'Routes to Diagnosis for cancer - Determining the patient journey using multiple routine datasets'. |
| cwt | The National Cancer Waiting Times Monitoring Data Set supports the continued management and monitoring of waiting times. |
| sact | Systemic Anti-Cancer Therapy (chemotherapy detail) data for cancer participants from PHE covering regimens between 03/2015 and 12/2018. One row per chemotherapy cycle, per tumour (SACT-specific pseudo_tumour_id), per participant. |
| rtds | The Radiotherapy Data Set (RTDS) standard (SCCI0111) is an existing standard that has required all NHS Acute Trust providers of radiotherapy services in England to collect and submit standardised data monthly against a nationally defined data set since 2009. The purpose of the standard is to collect consistent and comparable data across all NHS Acute Trust providers of radiotherapy services in England in order to provide intelligence for service planning, commissioning, clinical practice and research and the operational provision of radiotherapy services across England. Data is available from 01/04/2009. The data is linked at a patient level and can be linked to the latest available av_patient table. |
| ncras_did | The Diagnostic Imaging Dataset (DID) is a central collection of detailed information about diagnostic imaging tests carried out on NHS patients, extracted from local radiology information systems and submitted monthly. The DID captures information about referral source, details of the test (type of test and body site), demographic information such as GP registered practice, patient postcode, ethnicity, gender and date of birth, plus data items about different events (date of imaging request, date of imaging, date of reporting, which allows calculation of time intervals. Data is available for patients diagnosed between 1 January 2013 and 31 December 2015. |
| lucada_2013 | The National Lung Cancer Audit (LUCADA) looks at the care delivered during referral, diagnosis, treatment and outcomes for people diagnosed with lung cancer and mesothelioma. The data items in the LUCADA dataset have been compiled to meet the requirements of audit, and are not to be confused with the data items identified as Lung Cancer in the National Cancer dataset. The audit focuses on measuring the care given to lung cancer patients from diagnosis to the primary treatment package, assessing against standards and bringing about necessary improvements. The project supports the Calman Hine recommendations, the National Cancer Plan and other national guidance (e.g. NICE guidance) as it emerges. |
| lucada_2014 | As above. Different schema to lucada_2013. |
| sact_uncurated | This table extracts chemotherapy (SACT) information for cancer participants in the 100,000 genomes project from unlinked and unprocessed PHE/NCRAS chemotherapy data from 2008 until June 2020. Please refer to background and use caveats in the quality notes section of this release note. |
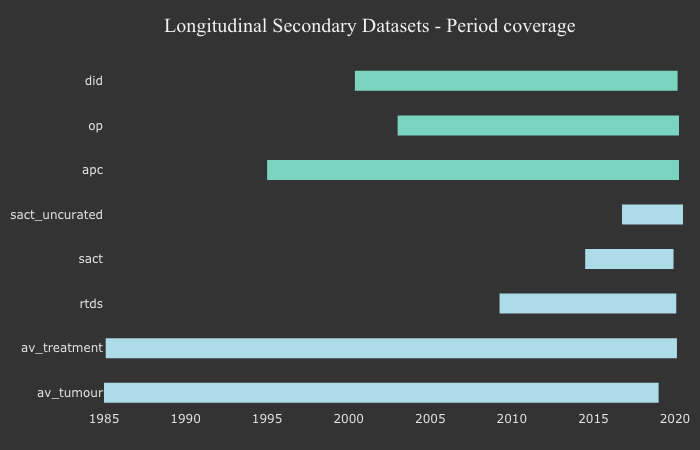
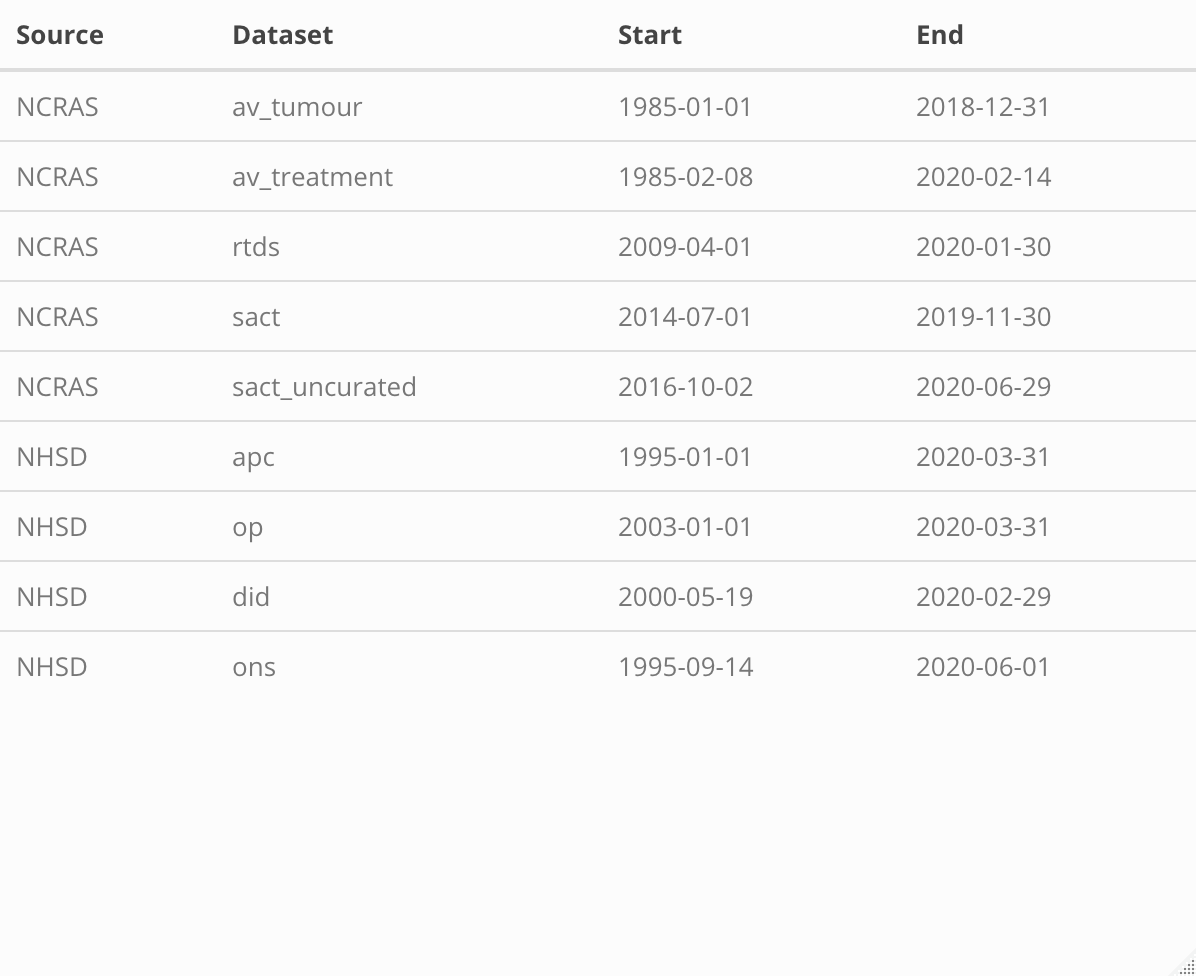
Activity Period Coverage for the longitudinal secondary data tables¶
Genomics England Data Resources¶
Genomics England data resources are available in the following locations:
From the AWS desktop:
~/gel_data_resources/
From the high performance compute (HPC) cluster:
/gel_data_resources/
The data resources available here are:
Tiering data for rare disease: Tiering data are available for rare disease participants who have been through the Genomics England interpretation platform. These data provide information on the pathogenicity of variants that have been identified in the proband’s genome. Tiering data for rare disease probands can also be found in the designated LabKey table outlined above (see tiering_data table in section 9.3). We have discontinued updating these data until further notice, and refer to the information provided in LabKey which is updated every release.
GMC exit questionnaires for rare disease: Outcomes questionnaire for interpreted genomes generated by Genomics England and Clinical Interpretation Providers (gmc_exit_questionnaire table in section 9.3). We have discontinued updating these data until further notice, and refer to the information provided in LabKey which is updated every release.
Interpretation request data for rare disease: The following information can be found within the interpretation request JSON file: Family Pedigree and Other Family History, Analysis Panels and versions, Specific Disorder, Tiered Variants and Tiering version, HPO terms, Workspace (NHS GMC or LDP site code), Gene Panel Coverage, Disease Penetrance, Variant Classification. We have discontinued updating these data until further notice, and refer to the information provided in LabKey which is updated every release.
Tiering, structural, and copy-number variant reports for Cancer: Annotated in JSON format. The file paths are available in the Quick View titled cancer_analysis.
Aggregated gVCF dataset (aggV2): This is a set of multi-sample VCF files containing germline genomic data from 78,050 participants – termed the “aggV2” – from Main Programme Data Release v10. This is an increase of genomic data of ~20,000 participant compared to our original aggregated data set (“aggV1”). The file contains germline samples from both the rare disease and the cancer programs including only genomes aligned to the Homo Sapiens NCBI GRCh38 assembly with decoys. All included samples have passed a set of basic QC metrics:
- Sample Contamination (freemix) < 0.03
- Ratio of SNV Het-to-Hom calls < 3
- Total Number of SNVs Between 3.2M-4.7M
- Array Concordance > 90%
- Median Fragment Size > 250bp
- Excess of Chimeric Reads < 5%
- Percentage of Mapper Reads > 60%
- Percentage of AT Dropout < 10%
These QC metrics are provided in the LabKey table aggregate_gvcf_sample_stats.
The aggregated dataset is split into 1,371 genomic regions or 'chunks' by physical position, to process the aggregation in parallel and to ensure that the resulting output files are not too large. No variant (= site) QC filters were applied in the dataset, but the VCF filter was set to PASS for variants which passed the following parameters:
- Missingness (fully missing genotypes with DP=0) ≤ 5%
- Coverage (Median Depth) ≥ 10X
- GQ (Median GQ) ≥ 15
- ABratio (Percentage of het calls not showing significant allele imbalance for reads supporting the ref and alt alleles) ≥ 25%
- completeGTRatio (Percentage of complete sites (sites with no missing data)) ≥ 50%
- phwe_eur (p-value for deviations from HWE in unrelated samples of inferred European ancestry) ≥ 1e-5
We recommend only using variants that have PASS in the filter column in your analyses which will have passed all the parameters above. If a site does not pass the parameters above, the failing criteria/criterion will be listed in the FILTER field in place of a 'PASS' flag, as detailed in the Site QC documentation.
In addition to the genotypes we provide pairwise kinship and relatedness information (PLINK2 implementation of the KING-Robust algorithm), principal components (of which the first 20 can be found in the aggregate_gvcf_sample_stats LabKey table), and predicted ancestry probabilities for all samples.
All data can be found at:
/gel_data_resources/main_programme/aggregation/aggregate_gVCF_strelka/aggV2/
Detailed documentation and examples for querying the data are available.
Our previous aggregated dataset (“aggV1”) containing germline genomic data from 59,464 participants can still be found at /gel_data_resources/main_programme/aggregated_illumina_gvcf/GRCH38/20190228/. However, we strongly recommend using the “aggV2” moving forward for all new analyses.
Cohort Metadata¶
Within the data release, there is genomic data and clinical data for participants that are part of non-NHS research cohorts that have been sequenced by Illumina and analysed via the Genomics England pipeline.
These research cohorts can be distinguished via their clinic ID as each has been given their own unique code. If any genomic or clinical data from the research cohorts is used in your analysis and subsequent publication, reference to the cohort organisation will need to be made.
| Non-NHS Cohort Name | Clinic ID | Rare Disease/ Cancer | Description | Constraints | Requirements of Use | Opportunities for further research |
|---|---|---|---|---|---|---|
| Breast Cancer Now | BCN | Cancer | The Breast Cancer Now Tissue Bank (BCNTB) is a multi-centre tissue bank established to fill the gap in the Triple Negative breast cancer (TNBC) research community. It systematically collects high quality tissues and data under an established ethical framework. Full clinico-pathological and follow-up data is due to be made available with ongoing longitudinal data collection. This cohort is curated group of 110 treatment naïve TNBC patients. Additional tissue for many is available through the BCNTB for further matched ‘omic analysis. |
Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | Potential to remove identifiers for the purpose of requesting access to Breast Cancer Now biobank samples. |
| CLL | CLL | Cancer | The original Chronic lymphocytic leukaemia (CLL) Genomics England Pilot aimed to develop the protocols and analytical methods required to perform whole genome sequencing (WGS) at scale for patients with CLL recruited into national clinical trials as a prelude to the Genomics England main programme. This cohort is a small subset of the pilot to allow for the provision of validation data. | Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | |
| UKALL2003 trial | ALL | Cancer | The aim of this project is to explore the genomic landscape of patients with acute lymphoblastic leukaemia at initial presentation in order identify mutations that could explain their poor response and potentially be future biomarkers. The objective was to perform whole genome sequencing and targeted screening for mismatch repair deficiency on a large well annotated cohort of patients with ALL treated on the UKALL2003 trial. This will generate, for the first time, a comprehensive genomic landscape of chemo-resistant acute lymphoblastic leukaemia. | Consistent with Genomics England acceptable uses | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data | |
| NIHR Bioresource | NB3 | Rare Disease | The NIHR BioResource is comprised of volunteers from around the country who have given their consent to taking of a biological sample, and they are willing to be approached to participate in research studies and trials on the basis of their genotype, and or phenotype. This cohort consists of rare disease participants who consented to WGS as part of the 100,000 Genomes Project. | Anyone who wishes to be granted permission to contact any of the NIHR BioResource participants should follow the process of applying to the NIHR BioResource. The steps to be made can be found on the [NIHR BioResource website]h(ttps://bioresource.nihr.ac.uk/about-us/about-the-bioresource/) | Any publication referencing the Sequence Data generated, needs to ensure reference is made to the contribution of the Provider to the generation of the Sequence Data |
Contact and Support¶
For all queries relating to this data release please contact the Genomics England Service Desk portal: Service Desk (accessible from outside the Research Environment). The Service Desk is supported by dedicated Genomics England staff for all relevant questions.
-
Some Rare Disease participants have multiple genomes, aligned to both GRCh37 and GRCh38. This excludes 86 TracerX genomes from 99 participants (refer to 6.4 for further information). ↩
-
Genomes which are yet to be classified as being rare disease or cancer are assigned an ‘unknown’ delivery type; therefore, the total cancer genomes + total rare disease genomes do not completely add up to the total genome count due to these ‘unknown’ delivery types. These genomes will be assigned to the rare disease or cancer programme at a later date. ↩