Exomiser¶
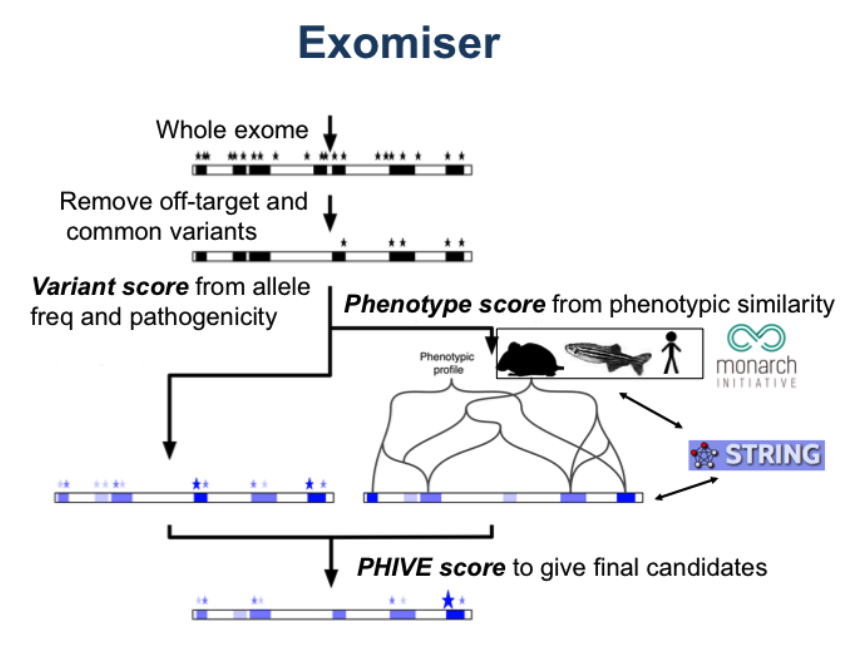
Exomiser is a programme that finds potential disease-causing variants from whole-exome or whole-genome sequencing data. All rare disease cases are run through the Exomiser automated variant prioritisation framework.
Given a multi-sample platypus VCF file, family pedigree and proband phenotypes encoded by Human Phenotype Ontology (HPO) terms, Exomiser annotates the consequence of variants (based on Ensembl transcripts) and then filters and prioritises them for how likely they are to be causative of the proband’s disease based on:
- the predicted pathogenicity and allele frequency of the variant in reference databases
- how closely the patient’s phenotypes match the known phenotypes of diseases and model organisms associated with the gene
Before the data is sent through the Exomiser pipeline, clinicians can manually review a case and add all the variants of a gene which has shown an association with the case disease to the list of variants to be analysed by Exomiser. In this scenario, even if variants of these manually added genes have an allele frequency higher than what Exomiser usually applies for variant filtering, these variants will still be annotated and included in the final table.
Variant interpretation¶
The variants are filtered based on the following criteria:
- Removing all low-quality and non-coding variants
- Compatibility with the modes of inheritance (MOI) being considered: autosomal dominant, autosomal recessive, x-linked dominant, x-linked recessive and mitochondrial
- Low frequency variants, <0.1% (or <2% for compound-heterozygotes) in all of the following reference databases: 100,000 Genomes Project reference samples, 1000 Genomes, ESP, TOPMed, UK10K, ExAC and gnomAD (excluding the Ashkenazi Jewish population).
Exomiser then calculates a score for how rare and pathogenic each variant is (on a scale of 0 to 1) using the above frequency sources and predicted pathogenicity scores by Polyphen2, SIFT and MutationTaster from dbNSFP.
For each MOI, the highest scoring compatible variant for each gene, or top two highest for compound-heterozygous candidates, are then selected as the contributing variant(s) for that gene under that MOI and used to assign a gene-level variant score (taking the mean for compound heterozygotes).
In parallel, Exomiser produces a phenotype score for each gene (on a scale of 0 to 1) based on how phenotypically similar the patient’s phenotypes are to:
- OMIM and Orphanet rare diseases known to be associated with the gene
- mouse and zebrafish models associated with the orthologue of the gene
- disease, mouse or zebrafish phenotypes associated with neighbouring genes in the StringDB protein-protein association database (scores weighted down based on network distance from the gene under consideration).
This scoring uses the OWLSim algorithm to semantically compare phenotypes such that similar but non-exact phenotypes can be identified and weighted according to how distant the two terms are in the ontology as well as how frequently observed is the phenotype in common. The highest score from these comparisons is assigned as the gene-level phenotype score.
Finally, a logistic regression model is used to combine the phenotype and variant scores and produce an overall Exomiser score for each gene and its contributing variants for each compatible MOI (scaled from 0 to 1).
A particular variant can be identified as contributing under a dominant MOI as well as a recessive MOI as a compound heterozygote and in this scenario will receive two different Exomiser scores. In this scenario, each MOIspecific score is returned as a separate reportEvent for that variant. The maximum Exomiser score out of any of the reportEvents for a variant is used to rank all of the returned variants with rank = 1 representing the most-likely candidate according to Exomiser and hopefully describing a rare, predicted pathogenic variant that disrupts a gene that has previously been associated with similar phenotypes to the patient.
You can learn more about the Exomiser algorithm in the Further reading and documentation section.
Location¶
Exomiser results for all available rare disease interpreted genomes can be found within the LabKey table, exomiser. Within this table, one can filter variants by: score (gene-phenotype, gene-variant, variant), variant rank, chromosomal position, gene name, variant consequence type, genotype, and phenotype, as well as other variables.
Validation¶
The Exomiser pipeline has been validated on 62 randomly selected, 100,000 Genomes Project cases with a positive diagnosis from the NHS GMCs (50 GRCh37 and 12 GRCh38).
The variant(s) reported as diagnostic by the NHS GMCs were correctly returned as the top ranked candidate(s) in 44/62 (71%) of cases (sensitivity = 0.71, precision = 0.71) and in the top five for 57/62 (92%) of cases (sensitivity=0.92, precision=0.18).
The five cases where the diagnosed variant lay outside the top five ranked Exomiser candidates included non-coding and non-penetrant diagnoses that Exomiser would not detect with the current pipeline.
Exomiser offers a complementary approach to the panel-based, tiering pipeline as shown below by an analysis of ~200 clinically solved cases. 72% of the diagnoses were identified in the applied gene panels by the tiering pipeline with high precision (1-2 candidates per case). Exomiser identified 81% of the diagnoses in its top five ranked results. Combining the tiering and Exomiser results leads to an increased recall of 90% of the diagnoses compared to using either approach alone, with a precision of 0.17, meaning an average of 5-6 variants are presented for consideration.