AggV3 code book - genotype queries¶
You can query the AggV3 genotype VCFs for participant genotypes. This document gives you example code for getting genotypes from all participants for a single variant, getting genotypes for a single participant across a genomic region and filtering variants by PASS sites.
You will find the genotype VCFs, organised by shard, at:
s3://357851407625-germline-aggregate-v3/data
There you will find folders for both the multiallelic and biallelic VCFs.
Querying the genotype VCFs requires the following steps:
- Identify the correct subshard for your analysis.
- Query the subshard VCF.
- Identify the source and type of the participants.
1. Identify the correct subshard for your analysis¶
There are two ways to identify the relevant subshards for your analysis:
- You can use the shard lookup tool to pull out the shards by inputting a locus.
- Query the shard BED files with bedtools. You can find the shard BED file for the biallelic VCFs at:
s3://512426816668-gel-data-resources/dragen3.7.8/AggV3_resources/manifests/genomic_data/biallelic_shards.bedAnd for the multiallelic VCFs at:s3://512426816668-gel-data-resources/dragen3.7.8/AggV3_resources/manifests/genomic_data/multiallelic_shards.bed
2. Query the subshard VCF¶
Now you can intersect query the subshard VCF in an interactive session or as a bash script. All the following queries use bcftools.
You will need to load bcftools in your terminal in your interactive session. You can do this easily using conda:
conda install bcftools
Filepaths
The following queries assume you have mounted only the relevant subshard VCF and index to your interactive session. If you have mounted the entire folder, you will need to modify the filepaths in the queries.
You will need to load bcftools as a container.
- Go to Batch analysis and select Run Pipeline.
- Search for bcftools and select a bcftools container
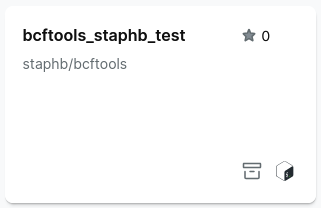
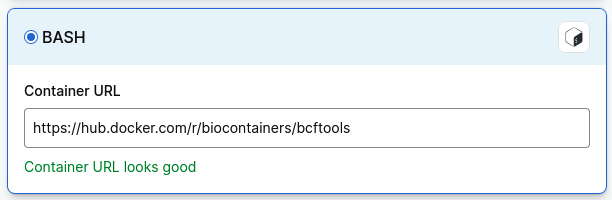
If you cannot find a bcftools container, select Import, then Bash and paste in the path to a bcftools container:
Extracting genotypes for a single variant¶
Question: I want to see the genotypes of all samples in aggV3 for a particular variant I know. For example the chr7: 48866984 - G/A SNP
Script: Use bcftools query. Enter the region (chromosome and position) using -r. The -f option formats the output with the attributes included. The > character writes the output to a tab-delimited file.
Select executable script and add the follow as a shell script:
#!/bin/bash
locus=$1
vcf=$2
output=$3
bcftools query -HH -r $locus \
-f '[%SAMPLE\t%CHROM\t%POS\t%REF\t%ALT\t%FILTER\t%GT\n]' \
$vcf > $output
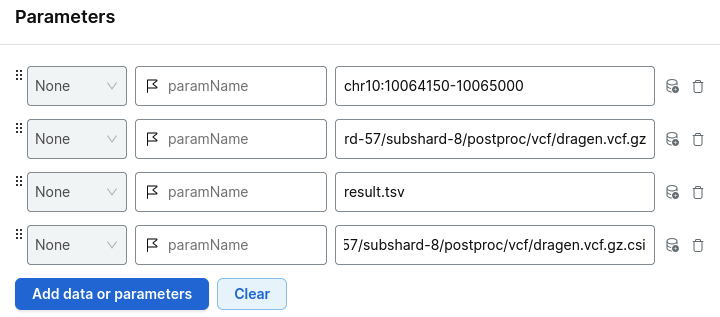
Add the parameters:
- your region of interest
- the relevant shard VCF file
- the index file
- your output file name
For example:
Choose your project and run analysis.
Output: The output is a tab-delimited file in long-format - where each sample is on a separate line for the same variant. The columns are in the same order as stated in the -f command above. If there are multiple variants at the same position (i.e. from a decomposed variant), all variants at the same position will be returned.
...
LP1234567-DNA_A01 chr7 48866984 G A PASS 0/0
LP2345678-DNA_B02 chr7 48866984 G A PASS 0/1
LP3456789-DNA_C03 chr7 48866984 G A PASS 1/1
LP4567890-DNA_D04 chr7 48866984 G A PASS 0/0
LP5678901-DNA_E05 chr7 48866984 G A PASS 0/1
LP6789012-DNA_F06 chr7 48866984 G A PASS 0/0
...
Data have been randomised and subset.
You can also extract additional per-sample FORMAT tags by wrapping the tags in the -f '[ ]' brackets of the bcftools query command. For example if you want to see the depth (DP) and genotype quality (GQ) for the variant above per sample, you could expand the -f command to:
Output:
...
LP1234567-DNA_A01 chr7 48866984 G A PASS 0/0 21 59
LP2345678-DNA_B02 chr7 48866984 G A PASS 0/1 28 81
LP3456789-DNA_C03 chr7 48866984 G A PASS 1/1 17 43
LP4567890-DNA_D04 chr7 48866984 G A PASS 0/0 36 79
LP5678901-DNA_E05 chr7 48866984 G A PASS 0/1 36 92
LP6789012-DNA_F06 chr7 48866984 G A PASS 0/0 26 56
...
You can also query a list of regions. You must use the -R flag and point to a tab delimited file of CHROM, POS, END.
Extracting genotypes for a single sample¶
Question: I want to see the genotypes of a particular sample in aggV3 for a region of interest. For example all variants for sample LP1234567-DNA_B10 in chr7: 48866084-48866984
Script: Use bcftools query. Enter the sample using -s and the region (chromosome and position) using -r. The -f option formats the output with the attributes included. The > character writes the output to a tab-delimited file.
Select executable script and add the follow as a shell script:
#!/bin/bash
platekey=$1
locus=$2
vcf=$3
output=$4
bcftools query -HH -s LP1234567-DNA_B10 \
-r $locus \
-f '[%SAMPLE\t%CHROM\t%POS\t%REF\t%ALT\t%FILTER\t%GT\n]' \
$vcf > $output
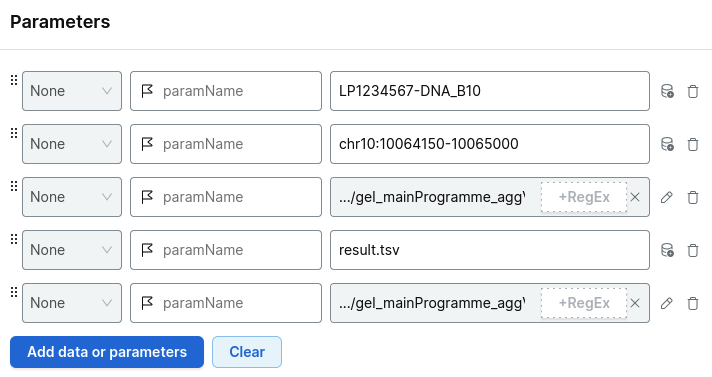
Add the parameters:
- your platekey of interest
- your region of interest
- the relevant shard VCF file
- the index file
- your output file name
For example:
Choose your project and run analysis.
Output: The output is a tab-delimited file in long-format - where each sample is on a separate line for the same variant. The columns are in the same order as stated in the -f command above.
...
LP1234567-DNA_B10 chr7 48866319 G T ABratio 0/1
LP1234567-DNA_B10 chr7 48866343 G A PASS 0/1
LP1234567-DNA_B10 chr7 48866346 A G PASS 1/1
LP1234567-DNA_B10 chr7 48866347 G A PASS 1/1
LP1234567-DNA_B10 chr7 48866357 A C PASS 0/0
LP1234567-DNA_B10 chr7 48866370 C A PASS 0/1
...
Data have been randomised and subset.
You can also query a list of samples. You must use the -S flag and point to a single column text file of sample IDs.
Filtering for PASS sites only¶
Question: "I only want to use PASS sites in my analysis".
Script: Use bcftools view. Use the -i option to set the FILTER to only include PASS sites. The -Oz option writes the output to a gzipped VCF and the -o option specifies the output VCF file name.
Select executable script and add:
bcftools view -i 'FILTER="PASS"'
Add the parameters:
-ryour region of interest- the relevant shard VCF file
-ooutput file- the index file
For example:
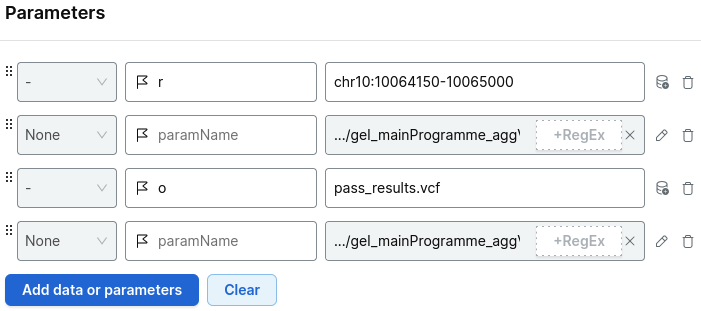
Output: Gzipped VCF of PASS sites only for the region of interest.
In order to query more detailed QC metrics, you will need to query our QC metrics VCFs.
3. Identify the source and type of the participants¶
For any further analysis, such as phenotype analysis, from the results of an AggV3 query, you will need to know the source of the samples. For this, we provide a sample list, available at:
s3://512426816668-gel-data-resources/dragen3.7.8/AggV3_resources/samples/sample_list/2026-01-23/sample_list_aggv3.csv
Sample list format¶
The sample list is a csv file
| Header | Description | Example |
|---|---|---|
participant_id |
pp1234567890 |
|
platekey |
LP1234567-DNA_A01 |
|
type |
rare_disease_germline, cancer_germline, covid_mild_germline or covid_severe_germline |
rare_disease_germline |
study_source |
GMS, MP100K or COVID |
GMS |
programme |
cancer, covid or rare_disease |
rare_disease |
participant_pmi_case_index |
Identifier to group participants which are linked to the same NHS ID. Deduplication process uses this identifier at its core. | 123456 |
programme_consent_status |
Consenting/Withdrawn. Consent status at programme (GMS/MP100K/COVID) level. Note that mid-release consent withdrawals may not accurately reflect the content of this column. | Consenting |
family_grouping |
Extended family group identifier. Example: In GMS if a family consists of two parents + two probands, these would be split in two trio-referrals. Referral A has parents + proband_A, and referral B has parents + proband_B. These two distinct referrals would be part of a single grouped family. | nhs_gms_family_001234 |
associated_case_reference |
gel_case_reference, or referral IDs associated with the participant's GMS case(s). Referral IDs are encrypted IDs and comma-separated where appropriate. | rr1234567890 |
dragen_delivery_id |
DR00123456 |
|
ploidy_estimation |
Sex karyotype ploidy estimates as provided by DRAGEN 3.7.8 output. | XX |
gold_platekey |
TRUE/FALSE. Part of the first 18,000 samples submitted for aggregation. These were selected for a variety of reasons and interests across research bioinformatics. We additionally selected samples with high amount of completeness for their meta-data (i.e. source library type). | FALSE |
aggV3_inclusion |
TRUE/FALSE | TRUE |
duplicate_of |
We have 18 duplicates (n = 36). This column provides the duplicated platekey of the duplicate pairing. | NA |
gvcf_path |
Path to the dragen gVCF file | s3://1234567890-realignment-dragen-3-7-8/100k_rd_analysis/pp1234567890/DR00123456/LP1234567-DNA_A01/LP1234567-DNA_A01.hard-filtered.gvcf.gz |
readSetARN |
readSetARN for the CRAM file in sequence store which was provided to Illumina for AggV3. | arn:aws:omics:eu-west-2:123456789012:sequenceStore/1234567890/readSet/2345678901 |
Query the sample list¶
To query the sample list, you should mount the sample list to your interactive session.
Let's assume you have queried AggV3 for genotypes with a locus and retrieved a tsv file called genotypes.tsv in the format:
...SAMPLE CHROM POS REF ALT FILTER genotype
LP1234567-DNA_A01 chr7 48866984 G A PASS 0/0
LP2345678-DNA_B02 chr7 48866984 G A PASS 0/1
LP3456789-DNA_C03 chr7 48866984 G A PASS 1/1
LP4567890-DNA_D04 chr7 48866984 G A PASS 0/0
LP5678901-DNA_E05 chr7 48866984 G A PASS 0/1
LP6789012-DNA_F06 chr7 48866984 G A PASS 0/0
...
You can intersect your genotype.tsv file against the sample file:
import pandas
sample_list = pandas.read_table('mounted-data-readonly/sample_list_aggv3_x.csv', sep=",", low_memory=False)
genotypes_list = pandas.read_table('genotypes.tsv', sep="\\t")
sample_genotypes = pandas.merge(genotypes_list, sample_list, left_on="#SAMPLE", right_on="platekey")[['CHROM', 'POS', 'REF', 'ALT', 'GT', 'platekey', 'participant_id', 'type', 'study_source']]
library(tidyverse)
sample_list <- read_csv('mounted-data-readonly/sample_list_aggv3_x.csv')
genotypes_list <- read_tsv('genotypes.tsv')
sample_genotypes <- merge(genotypes_list, sample_list, by.x="#SAMPLE", by.y="platekey")[,c('CHROM', 'POS', 'REF', 'ALT', 'GT', 'platekey', 'participant_id', 'type', 'study_source')]
You can now separate participants by study_source. You may also wish to filter by type.
Phenotype data accessibility
100kGP phenotype data can be accessed in both CloudOS and the main RE. You can choose whether to download the list to the VDI and query using LabKey, or to work with the data in CloudOS.
Covid phenotype data is only available in CloudOS.
NHS GMS data is only available in the main RE; you must download the list to the VDI and query using LabKey.
Take a look at our tutorial on getting medical data for participants.