AggV3 code book - querying QC metrics¶
Site QC data is provided in separate VCF files to the genotypes and functional annotation. These include information including depth, genotype quality, missingness, allele frequencies and genotype counts.
Querying the QC VCFs requires the following steps:
- Identify the correct subshard for your analysis.
- Query the subshard VCF.
1. Identify the correct subshard for your analysis¶
There are two ways to identify the relevant subshards for your analysis:
- You can use the shard lookup tool to pull out the shards by inputting a locus.
- Query the shard BED files with bedtools. You can find the shard BED file for the VCFs at:
s3://512426816668-gel-data-resources/dragen3.7.8/AggV3_resources/manifests/site_qc/2026-01-06/siteqc_shards.bed
2. Query the subshard VCF¶
Now you can query the subshard VCF in an interactive session or as a bash script. All the following queries use bcftools.
You will need to load bcftools in your terminal in your interactive session. You can do this easily using conda:
conda install bcftools
Filepaths
The following queries assume you have mounted only the relevant subshard VCF and index to your interactive session. If you have mounted the entire folder, you will need to modify the filepaths in the queries.
You will need to load bcftools as a container.
- Go to Batch analysis and select Run Pipeline.
- Search for bcftools and select a bcftools container
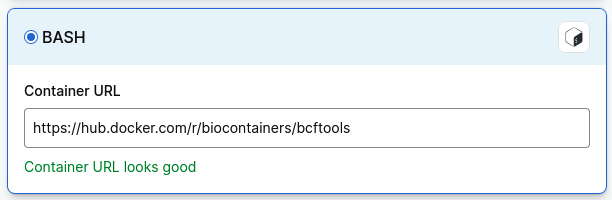
If you cannot find a bcftools container, select Import, then Bash and paste in the path to a bcftools container:
Extracting allele frequencies and site QC metics¶
Question: I want to see the allele frequencies of all variants in a region of interest. For example all variants in chr7: 48866084-48866984
Script: Use bcftools query. Enter the region (chromosome and position) using -r. The -f option formats the output with the attributes included. The > character writes the output to a tab-delimited file.
Select executable script and add the follow as a shell script:
#!/bin/bash
locus=$1
vcf=$2
output=$3
bcftools query -r $locus \
-f '['%CHROM\t%POS\t%REF\t%ALT\t%FILTER\t%INFO/AN\t%INFO/AC\t%INFO/AC_Hom\t%INFO/AC_Het\t%INFO/AC_Hemi\n]' \
$vcf > $output
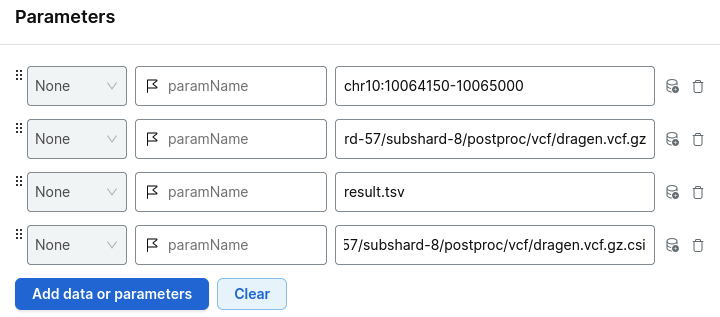
Add the parameters:
- your region of interest
- the relevant shard VCF file
- the index file
- your output file name
For example:
Choose your project and run analysis.
Output: The output is a tab-delimited file in wide-format - where each variant is on a separate line across all samples. The columns are in the same order as stated in the -f command above.
...
chr7 48866111 G C PASS 156390 222 1 222 0
chr7 48866116 A C PASS 156390 8 4 4 0
chr7 48866117 A T PASS 156390 12 2 2 0
chr7 48866118 T C PASS 156390 24 2 4 0
chr7 48866123 C T PASS 156390 110 4 10 0
chr7 48866128 G A PASS 156390 18 4 8 0
...
Data have been randomised and subset.
Allele frequency data for aggV3 are housed in the INFO field of the genotype VCFs as shown below:
##INFO=<ID=AN,Number=A,Type=Float,Description="Total number of alleles in called genotypes calculated across all samples.">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes calculated across all samples.">
##INFO=<ID=AC_Hom,Number=A,Type=Integer,Description="Allele counts in homozygous genotypes calculated across all samples.">
##INFO=<ID=AC_Het,Number=A,Type=Integer,Description="Allele counts in heterozygous genotypes calculated across all samples.">
##INFO=<ID=AC_Hemi,Number=A,Type=Integer,Description="Allele counts in hemizygous genotypes calculated across all samples.">
You can also extract per-variant summary statistics and site QC metics using bcftools query and including the INFO tag in the query. For example if you wanted to extract the per-variant median depth, median GQ and ABRatio, the -f command could be expanded to:
Output: