AggV3 code book - combining genotype and siteQC queries¶
You can query the AggV3 biallelic genotype VCFs for participant genotypes at variants of interest that meet quality control (QC) definitions. This pages describes how you can obtain genotypes for variants that pass QC within a specific region or gene of interest.
Querying for genotypes that meet QC criteria involves:
- Identify the correct genotype and siteQC VCFs for your analysis.
- Query the siteQC VCF.
- Filter the genotype VCF for passing variants.
1. Identify the correct genotype and siteQC VCFs for your analysis¶
There are two ways to identify the relevant subshards for your analysis:
- You can use the shard lookup tool to pull out the shards by inputting a locus.
- Query the shard BED files with bedtools.
2. Query the siteQC VCF¶
Now you can query the subshard VCF in an interactive session or as a bash script. All the following queries use bcftools.
You will need to load bcftools in your terminal in your interactive session. You can do this easily using conda:
conda install bcftools
Filepaths
The following queries assume you have mounted only the relevant subshard VCF and index to your interactive session. If you have mounted the entire folder, you will need to modify the filepaths in the queries.
You will need to load bcftools as a container.
- Go to Batch analysis and select Run Pipeline.
- Search for bcftools and select a bcftools container
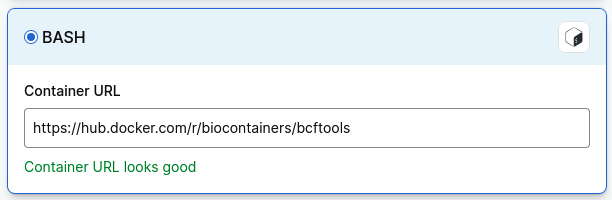
If you cannot find a bcftools container, select Import, then Bash and paste in the path to a bcftools container:
Filtering siteQC VCF for variants that meet QC criteria¶
The QC metrics provided in the siteQC VCFs are described in our SiteQC documentation.
Given you want to filter for variants in chr18:69196800-69196900 that pass the following criteria:
MEDIAN_DP>= 10MEDIAN_GQ>= 15MISSINGNESS_RATE<= 0.05AB_RATIO>= 0.25
Use bcftools to filter the siteQC VCF for variants that pass these thresholds, write out a list of variant IDs for the passing variants. Here, the variant ID is in the format CHROM:POS:REF:ALT to match the ID column present in the genotype VCF. The -i option applies the thresholds to the QC metrics. The -f option formats the output in the form CHROM:POS:REF:ALT. The -r filters for variants in the given genomic region.
bcftools query filesystems/dragen.gel.siteqc.vcf.gz -i '(MEDIAN_DP>=10) & (MEDIAN_GQ>=15) & (MISSINGNESS_RATE<=0.05) & (AB_RATIO>=0.25)' -r chr18:69196800-69196900 -f '%CHROM:%POS:%REF:%ALT\n' > siteqc_pass_variants.tsv
Select executable script and add the follow as a shell script:
#!/bin/bash
vcf=$1
locus=$2
output=$3
bcftools query $vcf -i '(MEDIAN_DP>=10) & (MEDIAN_GQ>=15) & (MISSINGNESS_RATE<=0.05) & (AB_RATIO>=0.25)' -r $locus -f '%CHROM:%POS:%REF:%ALT\n' > $output
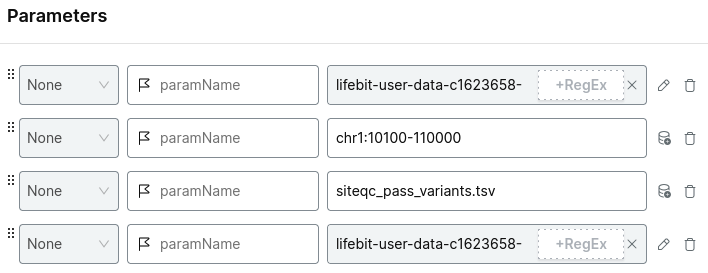
Add the parameters:
- the relevant siteqc shard VCF file
- the index file
- your region of interest
- your output file name,
siteqc_pass_variants.tsv
For example:
Choose your project and run analysis.
Output: The output is a single-column list of variant IDs for variants remaining after filtering.
chr18:69196801:G:GA
chr18:69196801:G:T
chr18:69196802:A:G
chr18:69196810:G:T
chr18:69196811:T:C
chr18:69196811:T:TCTA
chr18:69196814:A:C
chr18:69196819:C:A
chr18:69196819:C:T
chr18:69196824:A:G
...
3. Filter the genotype VCF for passing variants¶
The genotype VCF includes an ID column formatted as CHROM:POS:REF:ALT. You can use bcftools to filter variants by this ID, given a list of variant IDs in the same format.
Using the list generated in the previous step, you can filter the genotype VCF.
bcftools view -i 'ID=@siteqc_pass_variants.tsv' filesystems/dragen.vcf.gz -Oz -o pass_variants_filtered.vcf.gz
Select executable script and add the follow as a shell script:
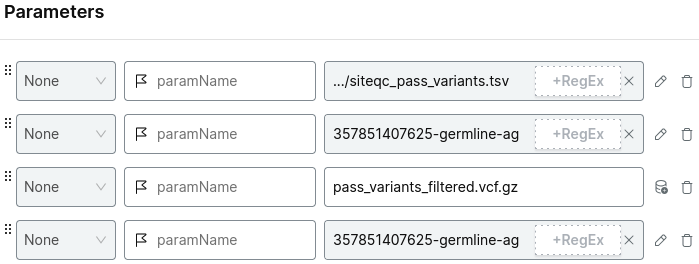
Add the parameters:
- the output file of the previous task
- the relevant genotype shard VCF file
- your output file name,
pass_variants_filtered.vcf.gz - the index file
For example:
Choose your project and run analysis.
Output: Gzipped VCF of variants within the region of interest passing the QC criteria.