Data generation, structure and locations¶
Data generation¶
Input data and data generation¶
The input data for AggV3 consisted of three elements that are part of our DRAGEN 3.7.8 data offering:
- Single sample *.cram
- Single sample *.hard-filtered.gvcf.gz.
- The estimated sex karyotypic ploidy estimates within the *-ploidy-estimates.csv file.
The gVCF then underwent a machine learning recalibration (MLR) step to reassess the confidence and probability of various variant calls. These MLR-gVCFs are available too, and were used as an input for the first step in the aggregation process.
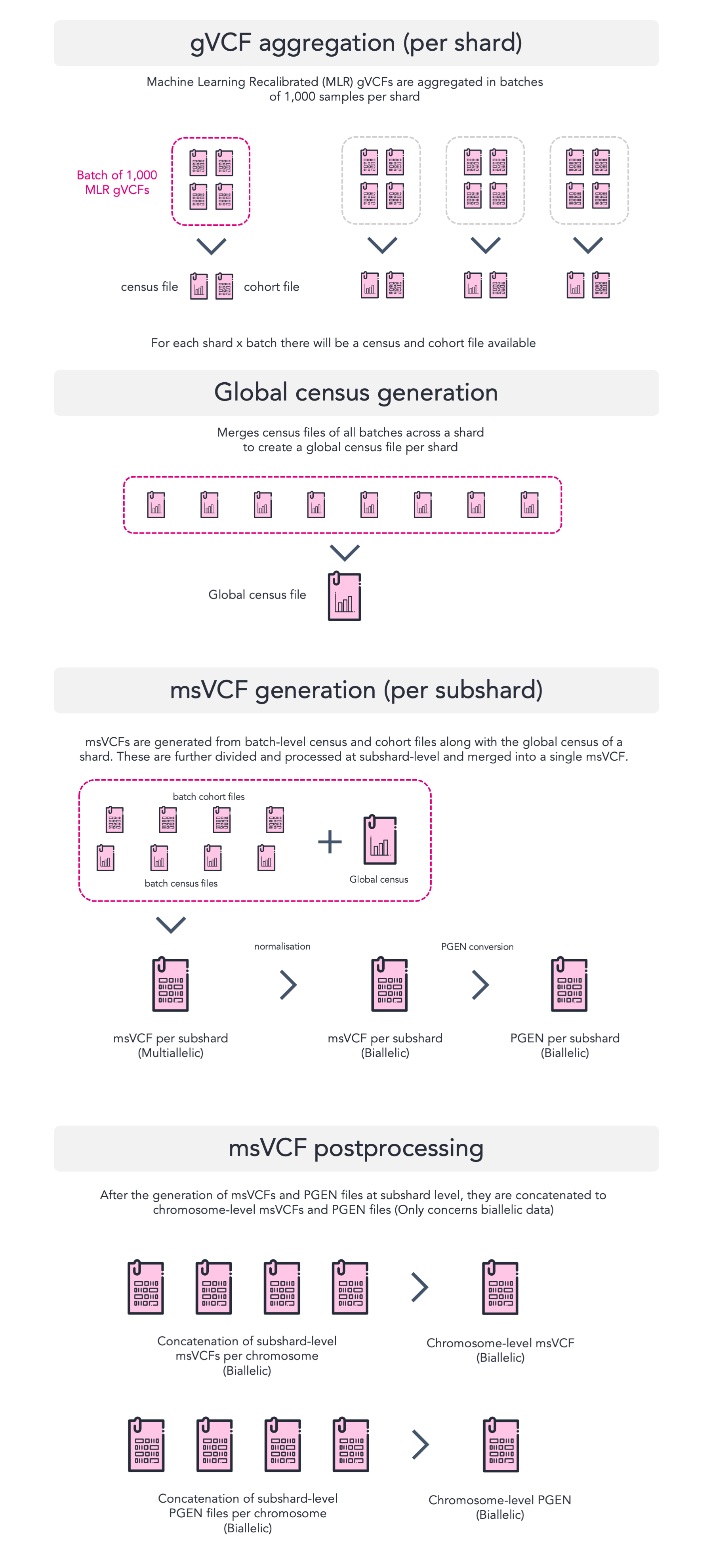
The aggregation process consists of a few steps which are explained in more detail in this blog post by Illumina. However, for the purpose of clarifying the process in the way that it was run for Genomics England, we have produced the figure below along with a short description. We do encourage you to read the blog post for further understanding.
Briefly, the various stages are as followed:
- gVCF aggregation: The aggregation of MLR gVCFs per 1,000 samples per subshard into a cohort file and a census file. The census file contains summary statistics of all variants and hom-ref blocks for each sample. The cohort file is akin to a condensed multi-sample gVCF.
- Global census generation: Generates a global census across all batches per subshard.
- msVCF generation; consisting of various sub-stages:
- Generates the multiallelic msVCF containing all genotypes and other format fields for all samples per subshard.
- The multiallelic msVCFs are then decomposed into a bi-allelic msVCF format per subshard containing genotypes only (along with INFO fields).
- The bi-allelic msVCFs are then used as an input for PLINK2 to generate PGEN files (PGEN, PSAM, PVAR).
- msVCF postprocessing: Concatenates all the bi-allelic output formats into chromosome-level files. For the multiallelic formats, only the sites and Illumina-provided annotation files are concatenated at chromosome-level as the msVCF itself would be too large in size.
For more detailed methodology information we refer you to the detailed methods section.
Data structure and types¶
The aggregate dataset consists of various different files and filetypes which may be relevant to your project in different ways.
Sharding¶
Because of the large size of an aggregate dataset, regions are split up in several msVCFs. Whereas AggV2 was previously split across >1,300 "chunks", AggV3 is split up by "shards" and "subshards".
AggV3 is divided in 102 shards, with each shard covering ~30 Mbp. These shards cover the autosomes, sex chromosomes, mitochondria, and ALT contigs. A single shard will never share more than one chromosome. Note that shard-16 is absent or empty as this covers the end of chromosome 2 which does not contain any variant.
| Chromosome Group | Shard Range |
|---|---|
| 1–22 | 1–93 |
| ChrX | 94–98 |
| ChrY | 99–100 |
| ChrM (Mitochondrial) | 101 |
| ALT contigs (e.g., chrUn, HLA) | 102 |
These shards are further divided into subshards. The amount can vary per shard, but in total there are 3,166 subshards (excluding the ALTcontig subshards, and shard-16). Most subshards have the maximum number of sites, which is 216,753 sites, but some have significantly less than that.
You can find the exact coordinates of the subshards in two locations:
- The shard lookup tool in our documentation.
- As BED files in S3 buckets within CloudOS, allowing you to use bedtools to identify the correct shard as part of your workflows. Our codebooks provide more details on how you would do this.
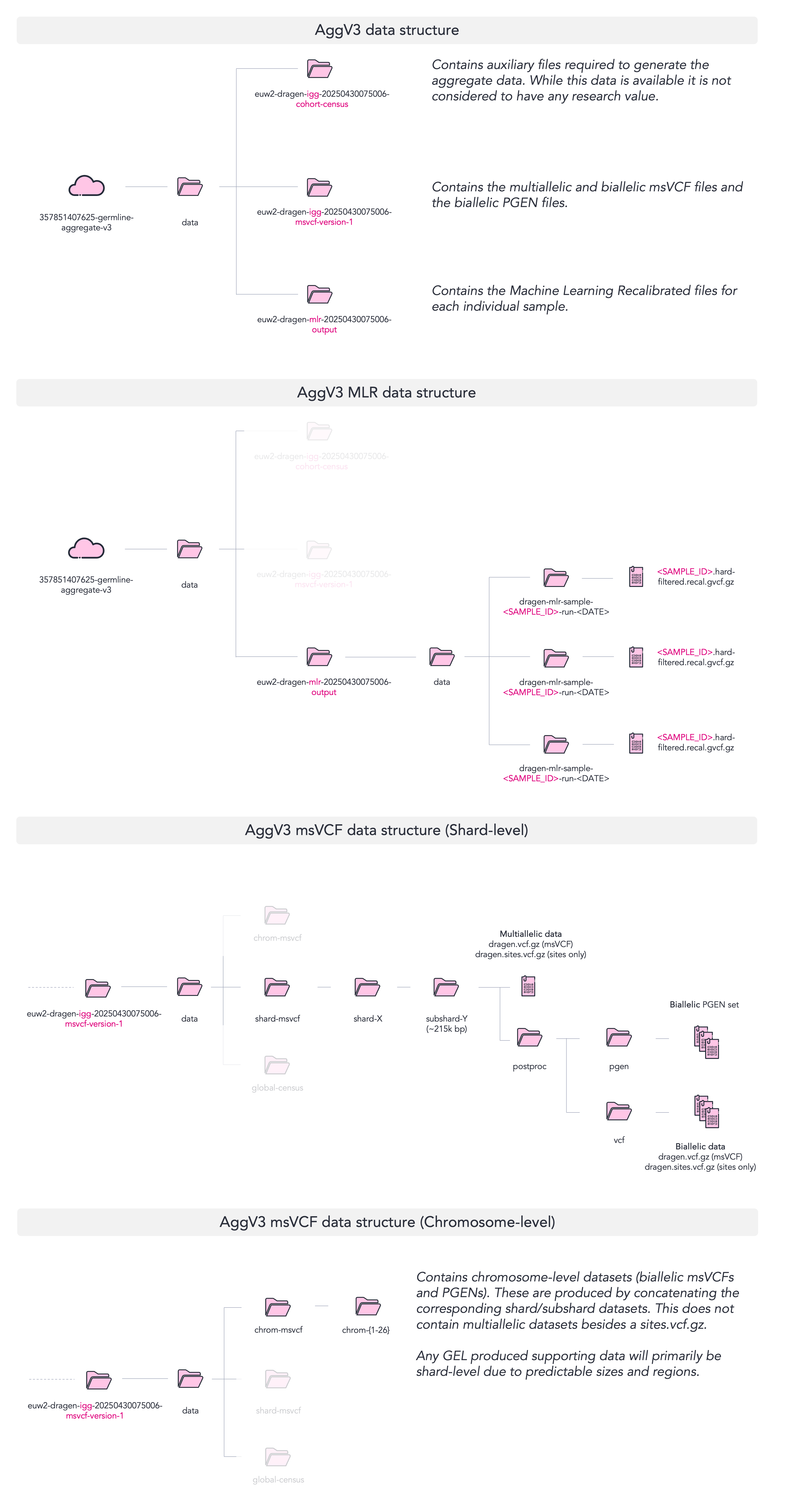
Data and file structure¶
We provide a consistent set of files across all subshards, with a consistent naming convention. Each subshard will contain a single multiallelic msVCF in its main folder (shard-msvcf/data/shard-*/subshard-*/dragen.vcf.gz) with a postprocessed (decomposed) bi-allelic msVCF in one of its subfolders (shard-msvcf/data/shard-*/subshard-*/postproc/vcf/dragen.vcf.gz).
This table provides details of all the files available. See the following section for a map to find a visual overview of where to find these files.
| Type | Region-type | Estimated size | Name | Location | Description |
|---|---|---|---|---|---|
| Bi-allelic msVCF | Subshard-level | < 5 GB | dragen.vcf.gz |
shard-msvcf/shard-\*/subshard-\*/postproc/vcf/dragen.vcf.gz |
Bi-allelic msVCF containing GT as the only format field. It also contains the QUAL, FILTER and INFO fields (AC, AN, AF, NS, NS_GT, NS_NOGT, NS_NODATA, LOW_CONF). |
| Bi-allelic site VCF | Subshard-level | ~ 5 MB | dragen.sites.vcf.gz |
shard-msvcf/shard-\*/subshard-\*/postproc/vcf/dragen.sites.vcf.gz |
Bi-allelic site VCF containing sites, QUAL, FILTER, and INFO columns – essentially the same info as the bi-allelic msVCF but without genotypes. Useful for faster position queries or existence of variants prior to obtaining samples with a variant of interest. For example, Genomics England annotations used these as input. |
| Bi-allelic PGEN files | Subshard-level | ~ 1 GB | dragen.pgen, dragen.psam, dragen.pvar |
shard-msvcf/shard-\*/subshard-\*/postproc/pgen/dragen.pgen<br>shard-msvcf/shard-\*/subshard-\*/postproc/pgen/dragen.psam<br>shard-msvcf/shard-\*/subshard-\*/postproc/pgen/dragen.pvar |
Bi-allelic PGEN files generated from the bi-allelic msVCF. These also contain the QUAL, FILTER and INFO columns from the bi-allelic msVCFs. |
| Multiallelic msVCF | Subshard-level | ~ 80–90 GB | dragen.vcf.gz |
shard-msvcf/shard-\*/subshard-\*/dragen.vcf.gz |
Multiallelic msVCF containing GT/GQ/LAD/FT/LPL/LAA/LAF/DP/QL/MQR as format fields. |
| Bi-allelic msVCF | Chromosome-level | ~ 100–250 GB | dragen.vcf.gz |
chrom-msvcf/chrom-\*/postproc-vcf/dragen.vcf.gz |
Chromosome-level bi-allelic msVCF containing GT as the only format field. This file was generated by concatenating the bi-allelic subshard-level msVCFs. |
| Bi-allelic site VCF | Chromosome-level | < 1 GB | dragen.sites.vcf.gz |
chrom-msvcf/chrom-\*/postproc-vcf/dragen.sites.vcf.gz |
Chromosome-level bi-allelic site VCF containing sites, QUAL, FILTER and INFO columns. This file was generated by concatenating the bi-allelic subshard-level site VCFs (CONFIRM). Useful for faster position queries or existence of variants prior to obtaining samples with a variant of interest. |
| Bi-allelic PGEN files | Chromosome-level | < 75 GB | dragen.pgen, dragen.psam, dragen.pvar |
chrom-msvcf/chrom-\*/postproc-pgen/dragen.pgen<br>chrom-msvcf/chrom-\*/postproc-pgen/dragen.psam<br>chrom-msvcf/chrom-\*/postproc-pgen/dragen.pvar |
Chromosome-level bi-allelic PGEN files. This file was generated by concatenating the bi-allelic subshard-level PGEN files. |
Sometimes the VCF headers contain INFO, FORMAT or FILTER fields that do not actually exist in the data. Please refer to the documentation for the valid fields.
Data organisation map¶
Below we provide a visual map to aid the navigation of the aggregate data. For chromosome-level data, you will find that there are msVCFs and PGEN filesets available, however these are only biallelic. Multiallelic msVCFs are only available at subshard-level. For this reason, and for predictability and scalablity, we primarily provide supporting datasets at shard- and subshard-level data.
Multiallelic vs biallelic¶
There are various differences between the multiallelic and biallelic msVCFs in AggV3:
- Multiallelic msVCFs have variants and all of their alternate alleles on a single row, whereas the biallelic msVCF has all the variants and their individual alleles represented on a single row.
- In the
FORMATfields, the biallelic msVCF only provides information on the sampleGTwhereas the multiallelic msVCF provides more complete sample level data for each site (i.e.GQandLAD). This was done in order to maintain a smaller storage footprint when using the files. However, there is no clear indication to tell whether a site is derived from a multiallelic site. This information will be included in our SiteQC files.
Please see the table below for a comprehensive view of the FORMAT fields present in the msVCFs:
| FMT Field | Biallelic msVCF | Multiallelic msVCF | Description |
|---|---|---|---|
GT |
Yes | Yes | Genotypes |
GQ |
No | Yes | Genotype Quality |
LAD |
No | Yes | Localised field: Allelic Depth. For multiallelic sites, informs the AD of REF, ALT1, ALT2. Never more than three comma-separated values; reference depth is always reported |
FT |
No | Yes | Sample FILTER |
LPL |
No | Yes | Local normalised, Phred-scaled likelihoods for genotypes as in original gVCF (without allele reordering) |
LAA |
No | Yes | Mapping of alt allele index from original gVCF to msVCF, comma-separated, 1-based (each value is the allele index in the msVCF) |
DP |
No | Yes | Approximate read depth |
QL |
No | Yes | Phred-scaled probability that the site has no variant in this sample (original gVCF QUAL) |
MLR |
No | Yes | Z-score from Wilcoxon rank sum test of Alt vs. Ref read mapping qualities |
Below is the summary of the INFO field:
The following metrics are provided by Illumina as a part of the DRAGEN pipeline. Please note that the metrics were calculated on all samples, regardless of their genetic ancestry. More details on each metric can be found in the DRAGEN v4.3 documentation. Please note this is not DRAGEN 3.7.8 documentation, and the metrics calculation may differ slightly between the DRAGEN versions.
| INFO Field | Biallelic msVCF | Multiallelic msVCF | Description |
|---|---|---|---|
AC |
Yes | Yes | Allele count in genotypes |
AN |
Yes | Yes | Total number of alleles in called genotypes |
NS |
Yes | Yes | Total number of samples |
NS_GT |
Yes | Yes | Total number of samples with called genotypes |
NS_NOGT |
Yes | Yes | Total number of samples with unknown genotypes (./.) |
NS_NODATA |
Yes | Yes | Total number of samples with no coverage |
IC |
No | Yes | The inbreeding coefficient |
HWE |
No | Yes | Exact conditional Hardy–Weinberg Equilibrium P‑value for each ALT allele |
ExcHet |
Yes | Yes | Exact conditional Excess Heterozygosity P‑value for each ALT allele |
HWEc2 |
No | Yes | Site‑wise chi‑squared Hardy–Weinberg Equilibrium P‑value |
ABHet |
No | Yes | Allele balance based on number of reads within heterozygous genotypes |
ABHom |
No | Yes | Allele balance based on number of reads within homozygous genotypes |
AF |
Yes | No | Allele frequency for each ALT allele |
Working with AggV3¶
To help you with working with this dataset on CloudOS, you can look at: