DRAGEN 3.7.8¶
Genomics England has realigned the vast majority of germline genomes from the 100,000 Genomes Project, NHS-GMS, and COVID-19 programmes with DRAGEN 3.7.8 to enable (germline) research without the need to adjust pipelines to different aligners and reference genomes. Each sample is run through our pipeline individually followed by a joint-calling process at family-level for small variants and CNVs.
Sample processing strategy¶
We have realigned the vast majority of the genomes available in the 100,000 Genomes Project, NHS-GMS, and COVID-19 programmes to DRAGEN 3.7.8, however there will be samples that have not been realigned. The primary factor for realignment was that the genomes were consented for research at that time, and selecting the latest delivery where multiple deliveries were present. A secondary factor would be the concordance between phenotypic and karyotypic sex as reported within the rare_disease_analysis and cancer_analysis tables for genomes of the 100,000 Genomes Project. Similarly, concordance between inferred sex and karytopic sex within our internal systems (openCGA) for NHS-GMS and COVID-19 genomes. If any sample within a family did not show concordance, the entire family was left out from realignment. And finally, for NHS-GMS genomes we currently have not realigned any families where no proband was available.
The concordance required between reported and karyotypic sex negatively affects cases recruited with a listed condition relating to sex development. These cases have been manually reviewed and curated by our Diagnostic Discovery team to provide a "meaningful" inferred sex for the DRAGEN aligner to take as an input. The DRAGEN pipeline requires an expectation on the number of expected X chromosomes within a sample where X0 and XY would be defined as male and XX (or alternatives) as female. A more defined sex karyotype is then further estimated by DRAGEN and provided within the ploidy.estimates.metrics.csv file which is further made available in the SampleQC output.
Realignment process¶
The realignment pipeline consists of multiple processes (also summarised on the figure):
- Mapping and variant calling
- Small variant joint-calling
- Merging gVCFs
- Joint variant calling
- CNV joint-calling
The DRAGEN 3.7.8 realignment pipeline runs mapping and variant calling within a single process taking a single BAM or CRAM file as an input. This process results in a new CRAM file and variant calls for small variants, structural variants (SVs), copy number variation (CNVs), count metrics, and much more. All of the outputs are available for researchers. This stage is then followed by joint-calling for small variants and CNVs using the pedigree file outlining the family structures. The joint genotyping stage for small variants will result in variants marked as denovo, however we did not specify the parameters to set quality thresholds for SNPs or indels to be considered as denovo so default values are used in this process. This means that denovo's can be retrieved (FMT/DN: 'Inherited', 'DeNovo', 'LowDQ') from the joint-called data. However, we encourage researchers to evaluate denovo's accordingly (see also FMT/DQ and FMT/DPL). Joint-calling for CNVs uses the tangent-normalised counts for each sample to generate joint-called CNV VCFs which should provide researchers with more accurate segmentation of CNVs.
For researchers interested in the CRAM files will find that these have been deep archived and inaccessible in the S3 bucket. After the realignment process, CRAM files are moved to and stored within AWS SequenceStore. Researchers will have access to an S3 alias or ARN (different type of address) which will act as a file path for their workflows and scripts.
For more details on DRAGEN 3.7.8 and its parameters, we refer you to either the online PDF, or the online documentation provided by Illumina. The command and parameters we used on our samples can be found within the VCF headers.

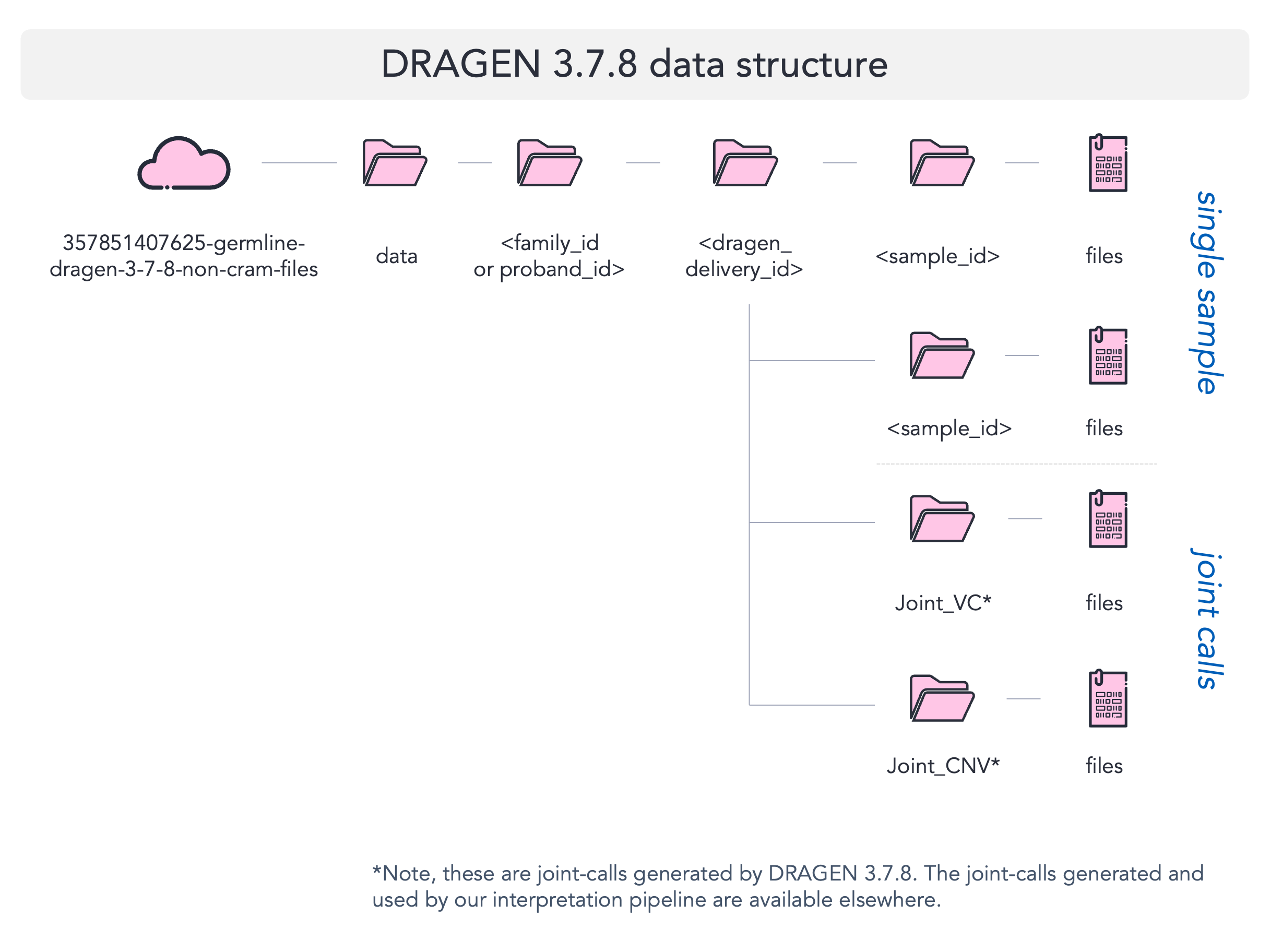
Data layout¶
DRAGEN 3.7.8 genomes are organised at family-level at a predictable level, meaning that within a family folder you will find individual folders per sample along with two folders containing the joint-calling results for small variants and CNVs. The provided delivery IDs are specific to the DRAGEN 3.7.8 and will not relate to any delivery ID associated with the original input data. Note that for genomes coming from the 100,000 Genomes Project family IDs are used for the parent directory whereas for NHS-GMS proband IDs are used for the parent directory.
`data/<family_or_proband_id>/<delivery_id>/
.../<sample_id_participant1>/
.../<sample_id_participant2>/
.../<Joint_CNV>/
.../<Joint_VC>/
| Type | Path |
|---|---|
| Single sample VCF | data/<family_or_proband_id>/<delivery_id>/<sample_id_participantX>/<sample_id_participantX>.hard-filtered.vcf.gz |
| Single sample gVCF | data/<family_or_proband_id>/<delivery_id>/<sample_id_participantX>/<sample_id_participantX>.hard-filtered.gvcf.gz |
| Single sample SV VCF | data/<family_or_proband_id>/<delivery_id>/<sample_id_participantX>/<sample_id_participantX>.sv.vcf.gz |
| Single sample SV VCF | data/<family_or_proband_id>/<delivery_id>/<sample_id_participantX>/sv/results/variants/diploidSV.vcf.gz |
| Single sample CNV VCF | data/<family_or_proband_id>/<delivery_id>/<sample_id_participantX>/<sample_id_participantX>.cnv.vcf.gz |
| Joint-called gVCF | data/<family_or_proband_id>/<delivery_id>/Joint_VC/<family_or_proband_id>_joint.hard-filtered.gvcf.gz |
| Joint-called CNV VCF | data/<family_or_proband_id>/<delivery_id>/Joint_VC/<family_or_proband_id>.cnv.vcf.gz |
We provide a table listing the paths to the VCFs generated from DRAGEN 3.7.8 and the corresponding CRAM file paths for all realigned samples. These tables are avaiable through CloudOS at s3://512426816668-gel-data-resources/dragen3.7.8/dragen378_resources/tables/dragen378_variant_call_paths.csv and s3://512426816668-gel-data-resources/dragen3.7.8/dragen378_resources/tables/dragen378_cram_paths.csv, respectively.
Data organisation map¶
Below we provide a visual map to highlight the folder structure for DRAGEN 3.7.8 data.