Cancer tiering¶
The 100kGP and NHS GMS cancer_tier_and_domain_variants table in LabKey lists variants of potential clinical significance in cancer. These could include oncogenic variants that destabilise protein function of a tumour suppressor, protective variants that disrupt an oncogene or variants of unknown significance. This allows you to query cancer-relevant genotypes.
The 100kGP and NHS GMS cancer_tier_and_domain_variants table in LabKey lists variants of potential clinical significance in cancer. These could include oncogenic variants that destabilise protein function of a tumour suppressor, protective variants that disrupt an oncogene or variants of unknown significance. This allows you to query cancer-relevant genotypes.
The table is based on the Gene centric SNV report for cancer participants. It is an aggregation of non-synonymous, splice site and RNA gene small variants found per sample for the the participants in the cancer programme.
The csv file generated by our internal cancer analysis team was used as a source and the paths of each participant's csv file are provided in the cancer_analysis Labkey table.
This table only contains domain and tiered variants, and is therefore not a complete picture of all variants in a sample. The majority of mutations included are somatic, and have been designated Domain 1, 2 or 3. Some germline variants occur in the table limited to those indicative of cancer predisposition as listed by Genomics England PanelApp. For information about how somatic and germline variants are interpreted, including how domain and tier assignment is done, please refer to sections 4 and 6 in the Cancer Genome Analysis Guide.
Variant annotation¶
Each variant carries information from Cellbase (cellbase_consequence) and Clinvar (clinical_significance) after annotation with ClinVar (version 2022-08-24 for 100kGP, version 2025-07 for NHS GMS). An assessment on variant loss of function has been included in the relevance column. Here a variant is marked as: loss of function (LoF) when the Cellbase consequence type matches those in Table 1, (likely) Pathogenic when the variant is present as such in ClinVar, and path_LoF when both are true. Remaining variants are marked as other.
| SO term | Consequence type |
|---|---|
SO:0001893 |
transcript ablation |
SO:0001574 |
splice_acceptor_variant |
SO:0001575 |
splice_donor_variant |
SO:0001587 |
stop_gained |
SO:0001589 |
frameshift_variant |
SO:0001578 |
stop_lost |
SO:0002012 |
start_lost |
SO:0001821 |
inframe_insertion |
SO:0001822 |
inframe_deletion |
Data overview (100kGP table)¶
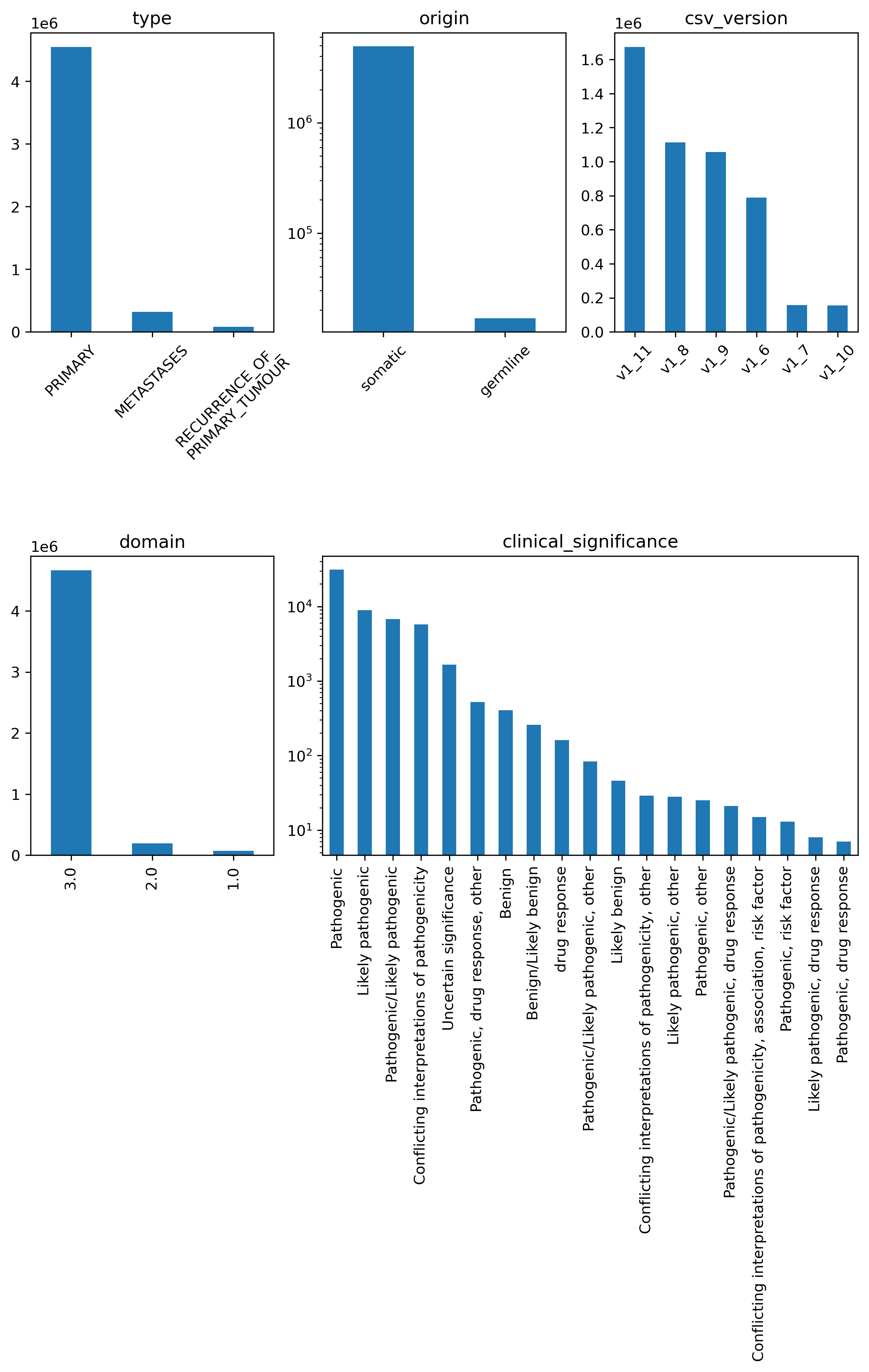
As of data release 16, the 100kGP version of this Labkey table is 5,001,535 rows long with 21 columns where each row represents a small variant within a tumour_sample_platekey. Value counts (>5) per: type, origin, domain and clinical_significance. These are on the total variants in the data, not normalised to the sample count. Please note the differing y-axis (log-scale or 1E6 modifier).
Data dictionary¶
Some columns are different between 100kGP and NHS GMS, these are indicated in the table.
| Field | Enumerations/Data Type | Description |
|---|---|---|
participant_id |
string | Participant Identifier (supplied by Genomics England). |
tumour_sample_platekey |
varchar | Concatination of Plate ID and Well ID - unique identifier for a proccessed well for tumour sample. |
disease_type |
string | The cancer type of the tumour sample submitted to Genomics England. Note: Some of the genomic analysis performed by the pipeline makes it possible to identify what cancer (disease type) the sample is from, and therefore correct potential errors in the disease type that was registered by the Genomic Laboratory Hub. As a result, the disease type in this table can be different from the disease type found in cancer_participant_disease. |
disease_sub_type |
string | The subtype of the cancer in question, recorded against a limited set of supplied enumerations. |
type |
string | sample type: PRIMARY, METASTASES or RECURRENCE_OF_PRIMARY_TUMOUR, as reported in .csv files. |
study_abbreviation |
string | TCGA study abbreviations, based on av_tumour histology and ICD10 codes for the given participant. For any participants where there is no data in av_tumour, the TCGA code was deduced from ICD10 codes in hes_apc. |
match_rank |
Enumerations:1 = Information in cancer_analysis, av_tumour and hes_apc all agree with one another.2 = Information in cancer_analysis and av_tumour agree 3 = Information in av_tumour and hes_apc agree4 = Information in cancer_analysis and hes_apc agree5 = No linkage - either there is no data in av_tumour or hes_apc, or there is no agreement between all three |
A categorical value describing the relationship between data in cancer_anaylsis, av_tumour and hes_apc |
origin |
string | the origin of a mutation differentiates between "somatic" and "germline" mutations. |
gene |
string | HUGO Gene Nomenclature |
transcript |
string | ENSEMBL transcript ID (ENST#) |
cellbase_consequence |
string | Cellbase consequence type |
change |
string | HGSV coding DNA reference sequence |
protein_change |
string | protein coding change |
chr |
string | chromosome, named as: 1-22, X, Y |
pos |
numeric | Position on chromosome (1-based) |
ref |
varchar | Reference Allele, sequence taken from the VCF file |
alt |
varchar | Alternate Allele, sequence taken from the VCF file |
domain |
Enumerations:1 = Domain 12 = Domain 23 = Domain 3 |
Domain 1: variants in a virtual panel of potentially actionable genes. Domain 2: variants in a virtual panel of cancer-related genes as curated by the Sanger's Cancer Gene Census. Domain 3: small variants in genes not included in domains 1 and 2. |
clinical_significance |
string | ClinVar clinical significance (version 2022-02-05) matched on gene and HGSV change. |
relevance |
string | LoF: the cellbase_consequence type confers protein loss of function. (likely) pathogenic: the entry has been annotated pathogenic or likely pathogenic by ClinVar. path_LoF: both the cellbase_consequence type confers loss of function and the variant is annotated as (likely) pathogenic by ClinVar. Other: Clinvar does not mark this variant as (likely) pathogenic, nor does it carry a mutation conferring protein loss of function. |
| Field | Enumerations/Data Type | Description |
|---|---|---|
participant_id |
string | Participant Identifier (supplied by Genomics England). |
tumour_sample_platekey |
varchar | Concatination of Plate ID and Well ID - unique identifier for a proccessed well for tumour sample. |
clinical_indication_full_name (NHS GMS only) |
string | Full name of clinical indication being tested for. |
type |
string | sample type: PRIMARY, METASTASES or RECURRENCE_OF_PRIMARY_TUMOUR, as reported in .csv files. |
origin |
string | the origin of a mutation differentiates between "somatic" and "germline" mutations. |
gene |
string | HUGO Gene Nomenclature |
transcript |
string | ENSEMBL transcript ID (ENST#) |
cellbase_consequence |
string | Cellbase consequence type |
change |
string | HGSV coding DNA reference sequence |
protein_change |
string | protein coding change |
chr |
string | chromosome, named as: 1-22, X, Y |
pos |
numeric | Position on chromosome (1-based) |
ref |
varchar | Reference Allele, sequence taken from the VCF file |
alt |
varchar | Alternate Allele, sequence taken from the VCF file |
domain |
Enumerations:1 = Domain 12 = Domain 23 = Domain 3 |
Domain 1: variants in a virtual panel of potentially actionable genes. Domain 2: variants in a virtual panel of cancer-related genes as curated by the Sanger's Cancer Gene Census. Domain 3: small variants in genes not included in domains 1 and 2. |
clinical_significance |
string | ClinVar clinical significance (version 2025-07) matched on gene and HGSV change. |
relevance |
string | LoF: the cellbase_consequence type confers protein loss of function. (likely) pathogenic: the entry has been annotated pathogenic or likely pathogenic by ClinVar. path_LoF: both the cellbase_consequence type confers loss of function and the variant is annotated as (likely) pathogenic by ClinVar. Other: Clinvar does not mark this variant as (likely) pathogenic, nor does it carry a mutation conferring protein loss of function. |
Help and support¶
Please reach out via the Genomics England Service Desk for any queries regarding the cancer tier and domain variants. We would welcome your feedback so that we can improve on our data offering.