Step-by-step guide to using containers¶
To use containers on the Genomics England HPC, follow the steps below:
- Identify the container you want to import
- Launch a job on the HPC
- Load Singularity
- Pull the container
- Mount files to the container
- Run the container
1. Identify the container you want to import ¶
On the HPC, you can pull in publicly available containers stored in dockerhub or quay.io. This can include containers you have built yourself and made public; you cannot pull containers that you have protected with credentials.
If you want to find publicly available containers, you may find a container listed with the documentation for the software you're using, or you may find what you need on a repository like biocontainers. Check for a dockerhub or quay.io container location on these websites.
The instructions for pulling a container from the original documentation (for example biocontainers) will not work in the RE. Please follow the instructions under Pull the container for how to pull.
2. Launch a job on the HPC ¶
You will need to launch a job on the HPC to use containers, and ensure you're using the full compute power of the HPC. For experimentation, we suggest using an interactive job. Once you have incorporated your container image into your pipeline, you may prefer to use a batch job.
3. Load Singularity ¶
Singularity 3.8.3 and 4.1.1 are available on the HPC. Load 4.1.1 with
module load singularity/4.1.1
Docker containers
Docker is not available on the RE, due to permissions issues with Docker. You can still use Docker containers with Singularity.
4. Pull the container ¶
To bring the container into the RE, you will need to use a pull command. This comprises:
singularity pull- The name for the singularity file that will be created. The file is named
filename.sif. You can call this whatever you choose. It will be saved to your current working directory. -
The web location of the container
For the location of the container, you will need to alter the URL in order to re-route by artifactory.
If you're using a dockerhub container in the form
container_path, you should change it todocker://docker-remote.artifactory.aws.gel.ac/container_path.If you're using a quay.io container in the form
quay.io/container_path, you should change it todocker://docker-quay-io.artifactory.aws.gel.ac/container_path.
It is possible to use run straight away, without first pulling the container, but no filename.sif file will be saved in your working directory.
Example pull commands¶
quay.io
Here we will pull deeptools from Biocontainers.
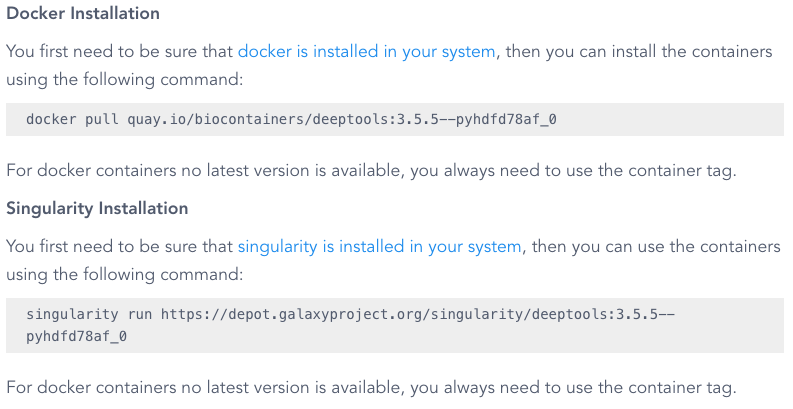
On the deeptools page, we can see a container location on quay.io, listed under "Docker installation":
quay.io/biocontainers/deeptools:3.5.5--pyhdfd78af_0
Even though there is also a command listed as "Singularity installation", we are not going to use this, since it uses a galaxy project repository location.
We will need to reroute this file location via artifactory, this changes it to:
docker://docker-quay-io.artifactory.aws.gel.ac/biocontainers/deeptools:3.5.5--pyhdfd78af_0
We can use this to pull with Singularity:
singularity pull my_deeptools.sif docker://docker-quay-io.artifactory.aws.gel.ac/biocontainers/deeptools:3.5.5--pyhdfd78af_0
dockerhub
Here we will pull bcftools from Dockerhub.
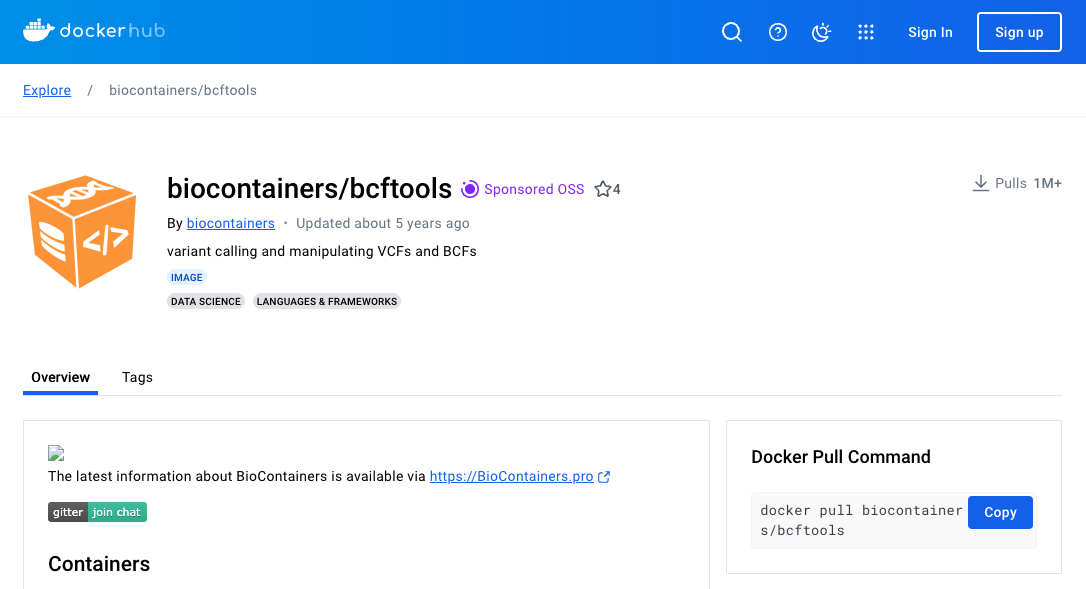
Here the pull location from dockerhub is listed as biocontainers/bcftools.
To reroute via artifactory, we will alter this to: docker://docker-remote.artifactory.aws.gel.ac/biocontainers/bcftools.
Our full pull command is:
singularity pull my_bcftools.sif docker://docker-remote.artifactory.aws.gel.ac/biocontainers/bcftools
5. Mount files to the container ¶
To analyse data with your container, you will need to mount or bind files to the container. This will allow the container to access the data. Do this using the --bind argument within your containers exec or run command.
relative paths
The paths you usually use to access folders in the RE are actually relative paths. When you mount paths to your container, you will need to use the full path. This means that to access the /genomes/ folder, you will need to use /nas/weka.gel.zone/pgen_genomes/.
To mount, you will need to --bind the full path, and then add another --bind for the relative path.
Some example binds¶
Include the relevant arguments in your run or exec commands. You can also bind specific files and folders within these listed folders:
-
genomes: containing the genomic VCF and alignment files
--bind /nas/weka.gel.zone/pgen_genomes:/nas/weka.gel.zone/pgen_genomes --bind /genomes:/genomes -
gel_data_resources: containing the results of Genomics England bioinformatics analysis including AggV2
--bind /nas/weka.gel.zone/pgen_int_data_resources:/nas/weka.gel.zone/pgen_int_data_resources --bind /gel_data_resources:/gel_data_resources -
public_data_resources: containing copies of public data
--bind /nas/weka.gel.zone/pgen_public_data_resources:/nas/weka.gel.zone/pgen_public_data_resources --bind /public_data_resources:/public_data_resources -
re_scratch: which you should use for all temporary files
--bind /nas/weka.gel.zone/re_scratch:/nas/weka.gel.zone/re_scratch --bind /re_scratch:/re_scratch -
re_gecip: the working folder for academic Research Network members
--bind /nas/weka.gel.zone/re_gecip:/nas/weka.gel.zone/re_gecip --bind /re_gecip:/re_gecip -
discovery_forum: the working folder for industry Research Network members
--bind /nas/weka.gel.zone/discovery_forum:/nas/weka.gel.zone/discovery_forum --bind /discovery_forum:/discovery_forum
6. Run the container ¶
There are two functions for running the software in a container: run and exec.
Many containers are written including an action, so they load the relevant software and then carry out the action. If you want to carry out that action, you should use run. For example:
singularity run \
--bind /nas/weka.gel.zone/re_gecip:/nas/weka.gel.zone/re_gecip --bind /re_gecip:/re_gecip \
my_container.sif \
/nas/weka.gel.zone/re_gecip/my_working_folder/my_file.tsv
If you wish to choose and run a particular function the software in your container, you should use exec. For example:
singularity exec \
--bind /nas/weka.gel.zone/re_gecip:/nas/weka.gel.zone/re_gecip --bind /re_gecip:/re_gecip \
my_container.sif \
function_in_container \
--file /nas/weka.gel.zone/re_gecip/my_working_folder/my_file.tsv