De novo variant research dataset¶
The de novo variant dataset contains all variants identified as de novo in 100kGP rare disease trios, where the variant found in the offspring cannot be identified in either parent.
The current release is v1, which was built upon the 100kGP V9 Data Release (released 02 Apr 2020). The dataset comprises genome-wide DNV annotation for 13,949 trios from 12,609 families from the rare disease programme.
Genome-wide DNV annotation was performed for all trios that have been successfully run through the Genomics England Rare Disease Interpretation Pipeline as of Data Release v9. The DNV annotation pipeline flags likely DNVs for each trio based on an array of filters that interrogate the multi-sample VCF outputs of the Platypus variant caller. The filters are grouped into two broad categories, base and stringent, and each variant is flagged if it fails any particular filter. We recommend using DNVs that pass the _stringent _filter in general, as these are more likely to be true DNVs.
Stringency
It is important to note that we adopt an inclusive approach by flagging likely DNVs in the VCF files without filtering any variants out. We provide all Mendelian inconsistencies in the VCF files for each family, together with a set of associated filters at the variant level, so that researchers can apply their own custom strategies to assess DNVs as required. At the end of this document, we supply code snippets and additional information to help users interrogate the DNV dataset.
The outputs of the DNV research dataset are:
| data | format | description |
|---|---|---|
denovo_cohort_information |
LabKey table | Cohort information for all participants included in the DNV dataset. Attributes within this table include: participant ID, sex, affection status, family ID, pedigree ID, and the path to each family's multi-sample VCF with flagged DNVs. |
denovo_flagged_variants |
LabKey table | All variants that pass base_filter for all trios within the DNV dataset. The table does not include variants that fail the base_filter due to size restrictions, but these can be found in the annotated multi-sample VCFs. This table includes all flags from the DNV annotation pipeline for each variant. |
| annotated multi-sample VCFs (family level) | VCFs | All multi-sample VCFs per family with DNVs flagged within the FORMAT field. These VCFs are functionally annotated with VEP and accessible within the filesystem. File paths per participant are included in the denovo_cohort_information LabKey table. The data can be found in directory: /gel_data_resources/main_programme/denovo_variant_dataset/ |
If you have any queries about the DNV dataset not covered in the FAQ at the end of this page, please contact us via the Genomics England Service Desk.
The de novo variant annotation pipeline¶
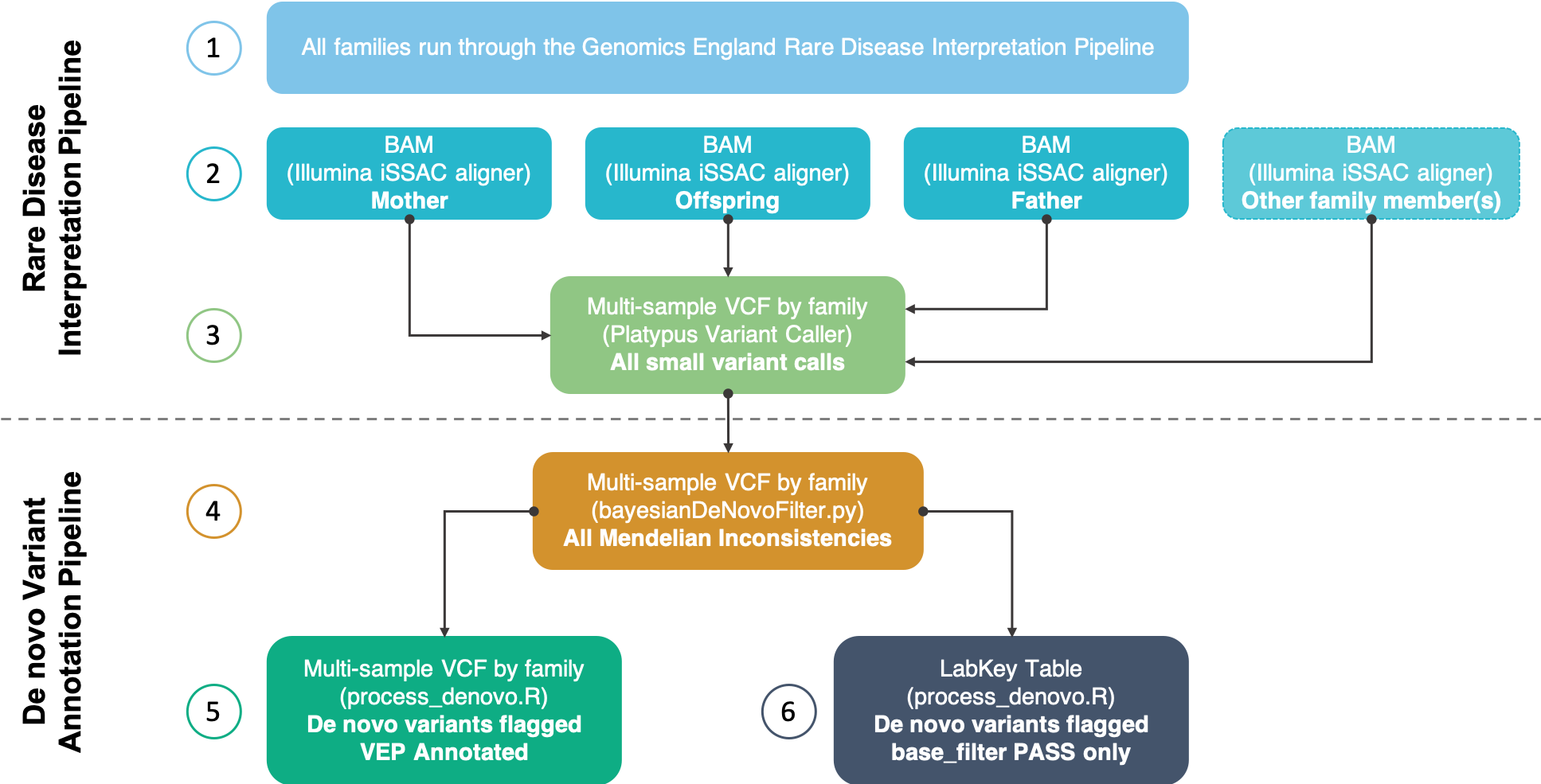
Rare disease interpretation pipeline¶
Step 1: Only families containing at least one trio that have successfully run through the Genomics England Rare Disease Interpretation Pipeline are considered in the DNV analysis.
Step 2: The BAMs for each member of the family are then fetched on the filesystem. The alignment is performed by the iSSAC aligner from Illumina.
Step 3: Small variants (SNPs and insertions/deletions up to 1,500bp) are jointly-called (and locally realigned) by family using the Platypus variant caller. This creates a multi-sample VCF for each family and contains the variants for every member of that family.
Nested trios
Note that some families contain additional members to the core trio such as siblings to the proband. We say that these families contain 'nested trios' as the DNV annotation pipeline will be run for each nested trio within a family.
De novo variant annotation pipeline¶
Step 4: For each family, the multi-sample VCF is fed into the Platypus bayesiandenovofilter.py python script. This script accompanies the Platypus variant caller. The pedigree and sex of each sample are also used as input. The script was run with default parameters - without changing the priors. More information can be found in the Additional Information section below. The output of this script is a multi-sample VCF with the same format as the input but only containing Mendelian inconsistencies for each offspring within the family.
Multi-sample VCF
In multi-sample VCFs containing nested-trios, Mendelian inconsistencies are reported per trio. Therefore, in a single variant line, one offspring may have a Mendelian inconsistency whilst the other may not.
Step 5: The custom DNV annotation script (process_denovo.R) is run separately by trio (i.e. nested trios within a family are run separately) using the multi-sample VCF as input. This script flags putative DNVs based on a set of filters shown below. The output of this script is a multi-sample VCF by family containing Mendelian inconsistencies for each offspring. The FORMAT column of the VCF is populated with the flags used to show evidence of the variant being a true DNV based on the filters shown below. Each VCF is then functionally annotated using Ensembl's Variant Effect Predictor (VEP).
Step 6: The VCF output for each family from Step 5 is then converted into a single data-frame and made accessible via LabKey. VEP annotation is not included in the LabKey table due to size restrictions (a single variant may have multiple genomic annotations due to the number of transcripts/isoforms).
The de novo variant annotation strategy¶
The DNV annotation strategy was built upon a combination of what is currently known in the literature and by advice and collaboration with members of Matt Hurles's group at the Wellcome Sanger Institute. We would like to thank Joanna Kaplanis and Patrick Short in particular (but any errors or omissions are our own). Initial checks suggest that the currently applied filters yield high specificity but may be filtering out some true de novo single nucleotide variants. We welcome all constructive feedback and any suggestions of how these filters could be modified in subsequent releases; please provide any such input via the Genomics England Service Desk.
The strategy is implemented in the custom R script (process_denovo.R) which is made available within the Research Environment. It comprises a series of four general filters that are grouped in to: Global, Base, Stringent, and Additional. These are used to flag each variant within a trio from a multi-sample VCF of Mendelian inconsistencies (Step 4). The flags for each variant within a trio are presented in the LabKey tables and within the annotated VCFs as described in the next section.
Global filters¶
Two global filters are firstly implemented. Beginning with the multi-sample VCF joint-called by family using Platypus, only Mendelian inconsistencies with the FILTER attribute set to PASS are considered. Additionally, only variants on the autosomes (1-22), the X-chromosome (X), and the mitochondrial chromosome (M) are considered (variants on scaffolds for example are excluded).
- PASS variants only
- Variants on chromosomes: 1 - 22, X, M
Base filters (autosomes)¶
Four filters are included in the base_filter category and aim to flag variants that are far more likely to be Mendelian inconsistencies than true DNVs.
Within a trio, if any of these four filters are marked as fail, then the base_filter is also set to fail.
Filters marked with an asterisk (*) are amended to account for variants on the X-chromosome as shown below.
| Filter | Pass Criteria | Description |
|---|---|---|
| zygosity_filter* | The genotype must be heterozygous (1/0 or 0/1) for the offspring and reference homozygous (0/0) in the mother and the father. | We only consider DNVs arising from a reference background. |
| mindepth_filter* | Minimum depth of 20X in the offspring and each of the parents. | This is taken from the NR FORMAT field in the VCF and ensures that DNVs have good coverage across all members of the trio. |
| maxdepth_filter | Maximum depth of 98X in the offspring. | Following findings from Rahbari et. al, 2016 |
| base_filter | All four individual base filters pass. | - |
Base filters (X-chromosome)¶
The two base filters below are adjusted to account for offspring sex and the X-chromosome. Please see the Additional Information section at the bottom of the page to understand how variants are coded on the X-chromosome by Platypus.
| Filter | Pass Criteria | Description |
|---|---|---|
| zygosity_filter | * Females, for PAR and non-PAR variants: The genotype must be heterozygous (1/0 or 0/1) for the offspring and reference homozygous (0/0) in the mother and the father. Males, for non-PAR variants: The genotype must be hemizygous (1) for the offspring and reference homozygous (0/0) in the mother. Males, for PAR variants:** The genotype must be hemizygous (1) for the offspring and reference homozygous (0/0) in the mother and the father. |
We only consider DNVs arising from a reference background. |
| mindepth_filter | * Females and Males, for PAR variants: Minimum depth of 20X in the offspring and each of the parents. Males, for non-PAR variants: Minimum depth of 20X in the offspring and in the mother. Females, for non-PAR variants: Minimum depth of 20X in the offspring and in the mother. Minimum depth of 10X in the father. |
This is taken from the NR FORMAT field in the VCF and ensures that DNVs have good coverage across all members of the trio. |
Stringent filters¶
An additional six filters are included in the stringent_filter category and aim to discriminate probable DNVs (that pass the base_filter) from high confidence DNVs. Variants have to pass the base_filter in order to be considered for the stringent_filter.
**Within a trio, if any of these six filters are marked as fail, then the stringent_filter is also set to fail. **
| Filter | Pass Criteria | Description |
|---|---|---|
| altreadparent_filter | No more than one read supporting the alternate allele in either the mother or the father. | This is taken from the NV FORMAT field in the VCF and ensures that there is minimal evidence of residual reads supporting the alternate allele in either parent. |
| abratio_filter | The AB ratio in the offspring is between 0.3 and 0.7. | The AB ratio is calculated by dividing the number of reads supporting the variant (NV) to the total number of reads at the variant site (NR). It ensures that the variant is not in allelic imbalance. |
| proximity_filter | DNV is not located within 20bp of another DNV within the same trio. | This only applies for variants that have already passed the base_filter. Therefore if two variants that pass the base_filter are within 20bp of one another within a trio, they are flagged as fail for the proximity_filter. |
| segmentalduplication_filter | No overlap with segmental duplications. | Taken from UCSC Table Browser for each genome assembly (Segmental Dups). Segmental duplications play an important role in both genomic disease and gene evolution. This track displays an analysis of the global organisation of these long-range segments of identity in genomic sequence. |
| simplerepeat_filter | No overlap with simple repeat regions. | Taken from UCSC Table Browser for each genome assembly (Simple Repeats). This track displays simple tandem repeats (possibly imperfect repeats) located by Tandem Repeats Finder (TRF) which is specialised for this purpose. |
| patch_filter | No overlap with patch regions. | Taken from UCSC Table Browser for each genome assembly (Fix Patches). When errors are corrected in the reference genome assembly, the Genome Reference Consortium (GRC) adds fix patch sequences containing the corrected regions. |
| stringent_filter | **All base and stringent filters pass. | - |
Additional flags¶
These additional flags are to aid downstream analysis. They are not used to inform the variant filters.
Flags marked as an asterisk are only available in the LabKey table _denovo_flagged_variants _and not in the annotated multi-sample VCFs.
| Filter ID | Description | Flags |
|---|---|---|
| snp_filter* | Flags whether the variant is a Single Nucleotide Polymorphism (SNP). | 0: Insertion/deletion; 1: SNP |
| in_par* | Flags whether the variant is in the pseudo-autosomal region (PAR). | 0: Not in PAR; 1: In PAR |
| gene_filter* | Flags whether the variant is in a coding exon (taken from GENCODE v32). | 0: In coding exon; 1: Not in coding exon |
| multidenovo_filter | No more than one identical Mendelian Inconsistency (by combining the chromosome, position, reference allele, and alternate allele) per trio within the same family. Only applicable for families containing nested trios. For families with nested trios (containing an additional sibling for example), if the same variant is determined to be a Mendelian Inconsistency in both siblings, this flag this is set to 0 (multidenovo). | 0: multidenovo; 1: not multidenovo |
Additional filters
Additional filters are not used as filters within the DNV annotation pipeline but can be used as a manual filter for downstream analysis.