Genomic data¶
For every sample we have the full output of whole genome sequencing (WGS) and analysis, carried out by Illumina, including alignments and called variants.
For a more in-depth description, you can consult the Whole Genome Sequencing Service Informatics and Cancer Analysis Services Guide from Illumina.
Different stages of the project used different sequencing and aligning technologies.
See how our genomic data was produced
Genomic data structure¶
Folders¶
There is a folder for each sample containing the genomic data for that sample. These data are an exact copy of those generated by the Illumina sequencing pipeline.
The sample folders are found in the following folder structure:
genomic: The main folder, accessible from both the desktop and HPC.by_date- Folders named by date of delivery in the form
yyyy-mm-dd- Delivery folders for each genome, in the form
HX00123456- Folders of genomic data, named for the sequencing platekey of the sample, which is a combination of plate barcode and well coordinate; for example:
LP12345678-DNA_A01
- Folders of genomic data, named for the sequencing platekey of the sample, which is a combination of plate barcode and well coordinate; for example:
- Delivery folders for each genome, in the form
- Folders named by date of delivery in the form
Below is an example of the genomic file structure for the sample LP3000987-DNA_A01:
| Genomes folder | By date folder | Delivery ID folder | Platekey folder |
|---|---|---|---|
| ~/genomes | ~/genomes/2018-04-25 | ~/genomes/2018-04-25/HX98765432 | ~/genomes/2018-04-25/HX98765432/LP3000987-DNA_A01 |
The same file structure is also accessible in s3 buckets using the CloudOS application. This is the only way to access genomic data for COVID-19 participants.
You can find the location of a participant's genome files using the latest LabKey genome_file_types_and_paths table or on the latest version Participant Explorer. All the participants you find by these methods are currently consented for use.
You should not try to find these files by directory traversing or browsing, as you may find genomes of participants who have since withdrawn consent and any requests to export these data via Airlock will be rejected.
In the desktop and terminal interface, you will be able see all genome delivery folders.
Note that this does not mean you will have access to all of these folders.
You will only have access to the genome folders that you have been given permissions to based on your credentials.
Files¶
The Illumina Whole Genome Sequencing Service and Cancer Analysis Service performs a series of processes with the following software packages.
All consented participants¶
| Software | Description |
|---|---|
| Issac | Aligns reads to the reference genome, trims and flags duplicates in the raw sequence. |
| Starling | A germline small variant caller and generates small variant (SNV and small indels ≤ 50 bp) analysis calls. |
| Manta | A germline and somatic structural variant caller; it generates structural variant (SV) analysis calls. |
| Canvas | A germline and somatic copy number variant caller; it generates copy number (CNV) and loss of heterozygosity (LOH) analysis calls. |
| Strelka | Joint tumour/normal small-variant caller. |
The tools used for alignment and variant calling will vary, particularly between 100K and NHS GMS data releases. For details, see the latest release information for 100K and NHS GMS
A subset of consented participants¶
| Software | Description |
|---|---|
| ExpansionHunter | A tool which looks for repeat expansions at several positions of interest. |
| HLATyper | A tools to generate likely HLA types for the sample. |
| ROHcaller | Identifies runs of homozygosity (ROHs) from whole-genome SNV variant call sets and predicts the most likely relationships of the sequenced individual's parents. |
The tools and versions used for each Illumina genome delivery will depended on the sample (germline/tumour) and on the Illumina pipeline version.
You can look at the header of the BAMs/VCFs to identify the Illumina pipeline and tool version used for each delivery.
All genomes have been aligned against either GRCh37 or GRCh38. For each data release, the reference genome is indicated in LabKey along with the path to the folder where the genome is stored.
Genomic data contents¶
An example file structure for LP12345678-DNA_A01 is shown below.
The key files are:
- ./Assembly/[Platekey].bam - Archival BAM file for sample
- ./Assembly/[Platekey].bam.bai - Index for the BAM file
- ./Variations/[Platekey].vcf.gz - Single nucleotide polymorphism (SNVs) and small insertion/deletion (1 bp–50 bp) calls in VCF format.
- ./Variations/[Platekey].genome.vcf.gz - Genome *.VCF file containing SNVs, indels, and reference covered regions
- ./Variations/[Platekey].SV.vcf.gz - Large Structural Variation calls (51 bp–10 kb) and copy number calls (10 kb+) in *.VCF format.
Note that the genotyping folders from early genome deliveries contain the results of the sample's run on the Infinium platform, an initial run done to confirm the sample identity and make sure that it is of high quality.
Quality metrics and other data such as coverage information can be found in the Metrics folder for each genome.
File types explained¶
BAM files¶
The BAM file contains all pass filter reads input into the analysis pipeline for a sample and includes aligned, duplicate and unaligned reads. It adheres to the SAM format specification wherever possible.
All vcf files are compressed and indexed using tabix; the tabix index files show up as the corresponding *.tbi file.
gVCF files¶
Human genome sequencing applications require sequencing information for both variant and nonvariant positions, yet there is no common exchange format for such data. gVCF addresses this issue.
gVCF is a set of conventions applied to the standard variant call format. These conventions allow representation of genotype, annotation and additional information across all sites in the genome, in a reasonably compact format (typically about 1/50 the size of the BAM file used for variant calling).
- gVCF is also equally appropriate for representing and compressing targeted sequencing results. Compression is achieved by joining contiguous nonvariant regions with similar properties into single 'block' VCF records. To maximise the utility of gVCF, especially for high stringency applications, the properties of the compressed block are conservative. Block properties such as depth and genotype quality reflect the minimum of any site in the block. The gVCF file is also a valid vCF v4.1 file and can be indexed and used with existing tools such as tabix and IGV.
Genomic data FAQs¶
Why can't I see all the genomes in the 'genomes/by_date' folder?
For some folders in the genomes/by_date folder, I receive the error: "Failed to open directory ...".
Why is this the case? Shouldn't I be able to see all genomes in that folder?
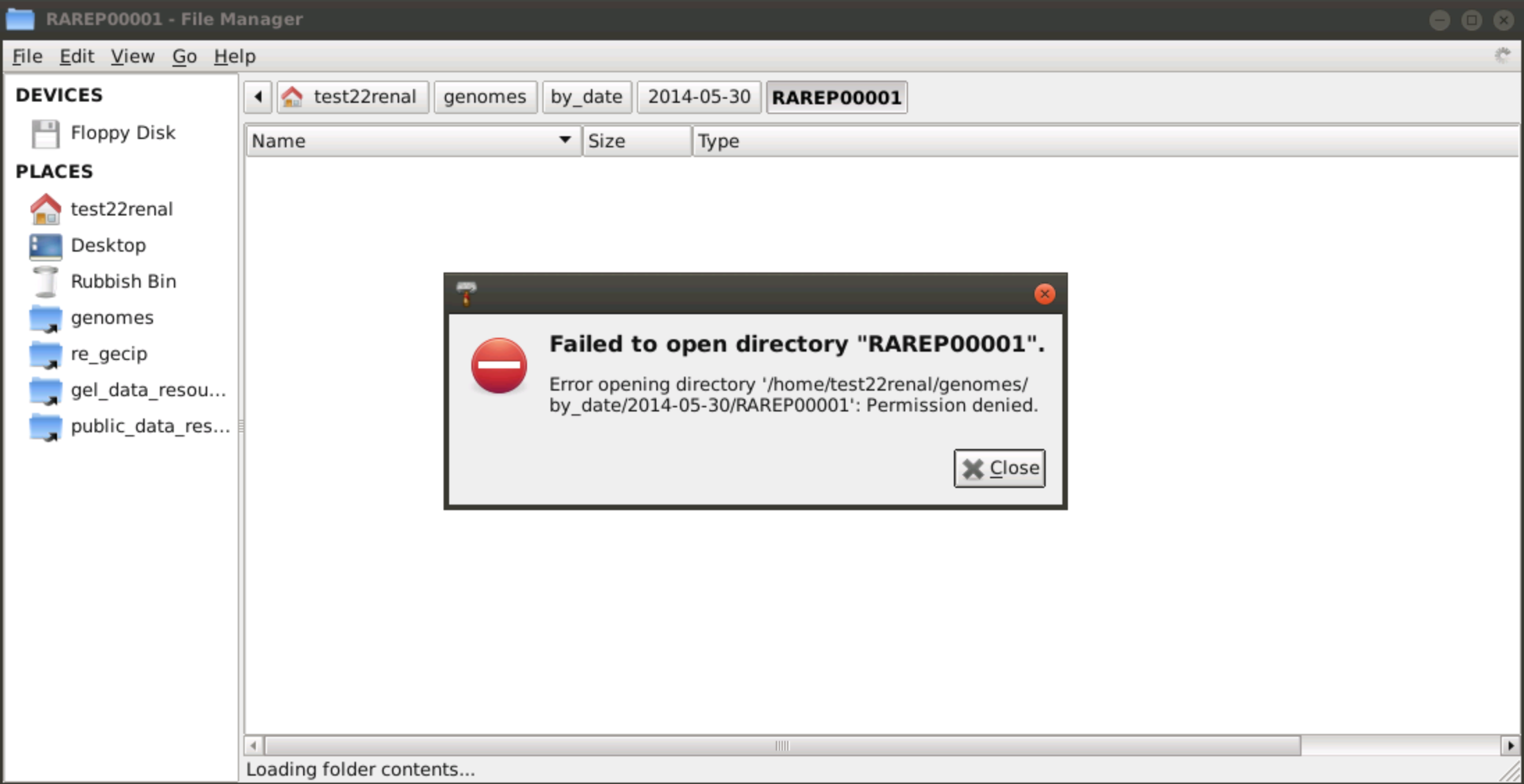
The genomes/by_date folder contains all genomes delivered to Genomics England by our sequencing provider. The overwhelming majority of these genomes are part of the 100kGP. As a Research Network member, you will have access to the 100kGP dataset as documented here. Some of the genomes however are not part of the 100kGP dataset and belong to sub-projects such as the Rare Disease Pilot or the Cancer 1K Cohort for example. You will not have access to these genomes as they are not part of the 100kGP. Also, as we release data into the Research Environment every three months, the latest genome deliveries will have not yet been provisioned. These will be accessible in a following 100kGP data release.
You can easily see which genomes you have access to by using the LabKey application on the desktop. Once signed into LabKey, select the 100kGP and then select the latest data release sub-project. In here, you will find a table called 'sequencing report'. This table contains the genome folders which are included in the 100kGP data release version you selected. If you are part of Research Network, you will have access to only these genome folders.
Why do some participants have multiple genomic data on the same reference assembly?
I have noticed that there are some individuals who have multiple genome alignments and VCF files on the same genome build (ie. one on GRCh37 and three on GRCh38). Why is this the case and is there an easy way to filter these out to just analyse one alignment per individual?
In the Research Environment, there are some participants who have multiple genome delivery IDs on the same reference assembly. Genome delivery IDs are assigned by Illumina and are related to an analysis run carried out in their system. Delivery IDs are unique and correspond to only one analysis run.
When a sample is re-delivered:
- The delivery ID will be exactly the same, if the original analysis has been re-delivered e.g. a BAM has not been transferred successfully and was truncated
- The delivery ID will be different, if Illumina re-run the analysis and therefore regenerated sample data
A re-run the Illumina analysis is often caused by one or more of the earlier genome deliveries not meeting the contractual or quality requirements. These include:
- Alignment File Quality Checks:
The BAM file contains the sequence read pairs for the sample mapped to the reference genome and is the source of all secondary data generated for downstream analysis such as somatic variants, SNVs, InDels and structural rearrangements. It is therefore important to check that this file is valid and formatted that it adheres to the SAM format. We check this by running a third-party software tool called ValidateSamFile from the Picard Toolkit. If the BAM is found to be invalid the pipeline will stop processing the sample and a fail status will be assigned to the run (Validate BAM Picard). Next, coverage distribution statistics are generated for the BAM file using samtools. These statistics are produced twice: once with base call and mapping quality filtering (Filtered Bamstats, Q30 Bamstats) applied and again with no filtering (Unfiltered Bamstats). The following base call and mapping quality filters are applied in the Filtered Bamstats step:
- exclude duplicates and secondary mappings
- exclude reads mapping outside autosomal non-N regions
- read mapping quality > 10
Samtools generates many metrics that can be used to evaluate the quality of the sequence alignment; these include: the number of mapped reads, average insert size, the number of read pairs mapped to different chromosomes (used to calculate the % chimeric DNA metric) and the number of duplicate reads.
The key statistics that are generated and automatically checked (by components Generate Q30 Metrics Bamstats, Generate Filtered Metrics Bamstats) and result in QC failure if they do not pass the threshold are:
| Metric Catalog Key | Metric Description | Threshold |
|---|---|---|
| perc_bases_ge_15x_mapQ_ge11 | bases in the genome covered by a read depth of at least 15X | ≥95% |
| GbQ30NoDupsNoClip | mapped bases with a base call quality of >=30 in gigabases [Gb] | 85Gb for germline samples 220Gb for somatic samples |
- Variant File Quality Checks:
The final part of the intake QC pipeline calculates statistics for the sample BAM and VCF files using the raw output generated by samtools and bcftools. VCF (variant call format) files containing variants are checked to ensure they are well formatted (VCF QC) and adhere to VCF 4.1 specifications. This is done using bcftools and gnuplot. Statistics generated by bcftools include the number of SNPs in the VCF, number of InDels and number of multiallelic SNP sites. The full list of stats generated for the VCF is given in Appendix 3. Any uncompressed VCF files are compressed and indexed using bgzip and tabix (part of the samtools).
Once genome deliveries have passed intake QC, they are subjected to further data quality checks to determine whether the samples are appropriate for downstream analysis and to prepare the data for interpretation. Again, if a a genome delivery fails at any one of these stages, a re-run of the Illumina pipeline will be requested and a subsequent genome delivery sent.
Reference genomes within the Research Environment
For my script, I am required to use a reference genome as an input. As we cannot download anything in the Research Environment, does Genomics England provide a local version of these reference genomes? And if so, where can I find these?
Yes, we indeed have local versions which can be found in /public_data_resources/.
For GRCh37 (V2):
/public_data_resources/reference/GRCh37/Homo_sapiens.GRCh37.75.dna.primary_assembly.fa
For GRCh38 (V4):
/public_data_resources/reference/GRCh38/GRCh38Decoy_no_alt.fa
For GRCh38 (DRAGEN):
/public_data_resources/reference/GRCh38DeAlt_HLA/GRCh38_full_analysis_set_plus_decoy_hla.fa