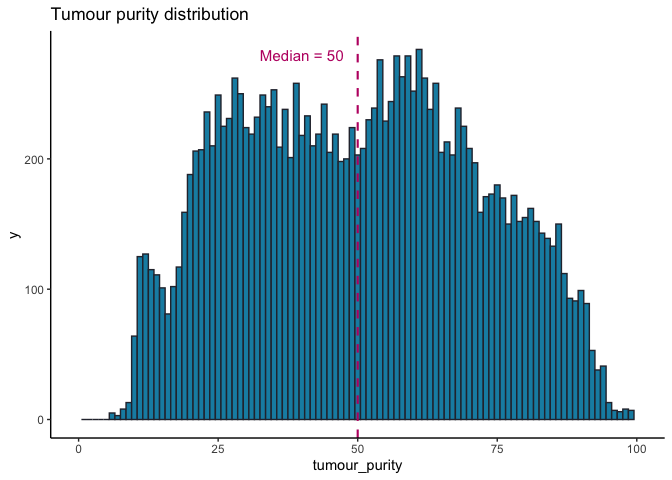
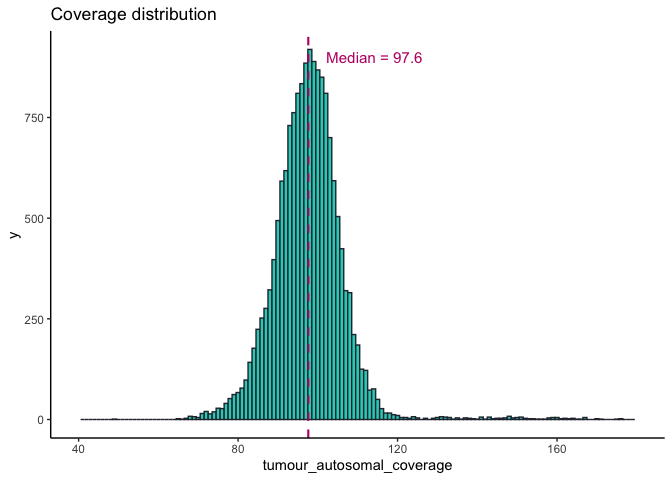
somAgg sample stats¶
There are 16,341 samples present in the somAgg. These include all samples that are present in cancer_analysis on the 100kGP Release v12 and are all the somatic samples that were successfully sequenced and interpreted.
Single sample sequencing and variant calling¶
somAgg combines the annotated somatic VCF files generated by Strelka and annotated with CellBase. Each tumour sample has a matched germline, both deep whole-genome sequenced with an average coverage of 100x and 30x, respectively. Few patients had more than one tumour sample sequenced.
| Step | Method |
|---|---|
| Preparation | Illumina TruSeq DNA Nano, TruSeq DNA PCR-Free or FFPE library preparation kit |
| Sequencing | HiSeq X generating 150 bp paired-end reads |
| Primary WGS analysis | Illumina’s North Star pipeline (version 2.6.53.23) |
| Read alignment | against human reference genome GRCh38-Decoy+EBV with iSAAC Aligner (version iSAAC- 03.16.02.19) |
| Small variant calling together with tumour-normal subtraction | Strelka2 (version 2.4.7) |
Strelka FILTERs flag the following germline variant calls as NOT PASS, they are nonetheless included in the single vcf files and somAgg:
- All calls with a sample depth three times higher than the chromosomal mean
- Site genotype conflicts with proximal indel call. This is typically a heterozygous SNV call made inside of a heterozygous deletion
- Locus read evidence displays unbalanced phasing patterns
- Genotype call from variant caller not consistent with chromosome ploidy
- The fraction of basecalls filtered out at a site > 0.4
- Locus quality score < 14 for for het or hom SNP
- Locus quality score < 6 for het, hom or het-alt indels
- Locus quality score < 30 for other small variant types or quality score is not calculated
Single sample decomposition¶
The annotated small variant VCF files used as input have been decomposed. The annotated single VCF files are generated from the somatic small variant VCF files (somatic_small_variants_vcf_path in cancer_analysis) generated by the variant calling pipeline, which comprises Strelka2 and vt for the decomposition. In other words, the somatic variant VCF files are the ones decomposed, which means that no multiallelic entries are found, because each multiallelic is represented by two or more bi-allelic variants.
The decomposition procedure is done in three steps by vt as presented here:
- Decompose variants of the same length.
vt decompose_blocksub -p {vcf_input} -o {vcf_output}
- Split records with multiple alternate alleles into multiple bi-allelic records an e.g. 1/2 genotype will be split to 1/. and ./1. The flag -s (“smart”) option makes INFO and FORMAT fields of type A and R to be retained and decomposed appropriately.
vt decompose -s {vcf_input} -o {vcf_output}
- Left-align indels and trim redundant bases. The “non-ambiguous” reference genome is used. This file only contains A,T,G,C and N characters.
vt normalize -n -w {window_size} -r {reference} {vcf_input} -o {vcf_output}
Genotype-level metrics¶
All 16,341 samples included in somAgg have successfully passed our internal sequencing and interpretation pipeline. These sample are listed in the LabKey table cancer_analysis. Some quality control statistics for these samples are provided below.
| Sample Attribute | Description |
|---|---|
| Tumour Cross-Contamination | less than 5% |
| Germline Cross-Contamination | less than 3% |
| Median Fragment Size | greater than 279bp |
| Excess of Chimeric Reads | mean of 0.3% |
| Percentage of Mapped Reads | mean of 93.4% |
| Percentage AT Dropout | mean of 3.1% |
Sample source and library preparation¶
The vast majority of the samples has been collected using surgical resection.
| tissue_source | number_of_samples | percent_of_samples |
|---|---|---|
| SURGICAL RESECTION | 14602 | 89.36 |
| NOT SPECIFIED | 521 | 3.19 |
| USS GUIDED BIOPSY | 490 | 3.00 |
| ENDOSCOPIC BIOPSY | 227 | 1.39 |
| NON GUIDED BIOPSY | 136 | 0.83 |
| BMA TUMOUR SORTED CELLS | 133 | 0.81 |
| NON STANDARD BIOPSY | 85 | 0.52 |
| CT GUIDED BIOPSY | 69 | 0.42 |
| STEREOTACTICALLY GUIDED BIOPSY | 49 | 0.30 |
| ENDOSCOPIC ULTRASOUND GUIDED FNA | 12 | 0.07 |
| LAPAROSCOPIC EXCISION | 8 | 0.05 |
| MRI GUIDED BIOPSY | 6 | 0.04 |
| ENDOSCOPIC ULTRASOUND GUIDED BIOPSY | 3 | 0.02 |
Also, the majority (~92%) of somAgg are from fresh-frozen (FF) and (~88%) from PCR-free.
| library_type | preparation_method | number_of_samples | percent_of_samples |
|---|---|---|---|
| PCR-Free | FF | 13711 | 83.91 |
| PCR | FF | 1285 | 7.86 |
| PCR | FFPE | 602 | 3.68 |
| PCR-Free | EDTA | 494 | 3.02 |
| PCR-Free | ASPIRATE | 124 | 0.76 |
| PCR-Free | CD128 SORTED CELLS | 70 | 0.43 |
| PCR | CD128 SORTED CELLS | 25 | 0.15 |
| PCR | EDTA | 19 | 0.12 |
| PCR | ASPIRATE | 8 | 0.05 |
| PCR-Free | FFPE | 3 | 0.02 |
As expected, we see an increased AT drop-out for FFPE samples, but overall the vast majority of samples have good mapping rate.
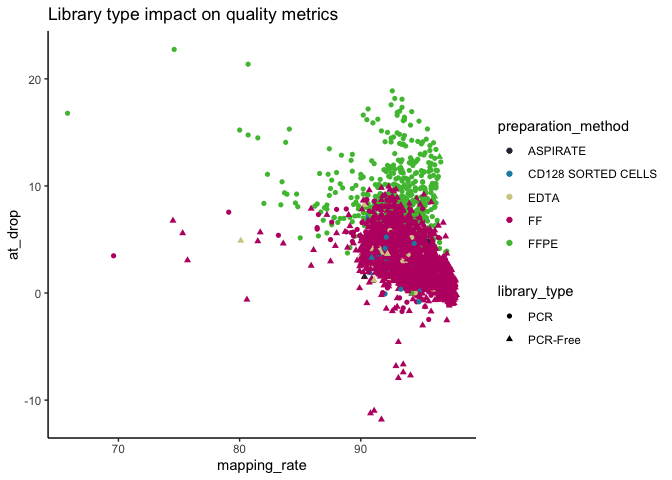
Tumour purity and coverage¶