SVCNV version 1.2¶
This workflow extracts large structural and copy number variants the specified gene region(s) or input regions from Illumina Structural Variant VCF files for either germline or somatic cohorts.
Description¶
While most of the research is focussed on small variants, large structural variants (SVs) are often overlooked. Here we provide a workflow that focusses on the extraction of SVs from our cohort data. This is the sister workflow to the genevariantWorkflow.
The workflow is written in WDL and is run using the Cromwell engine - version 1.0. It relies on four external tools in order to execute the workflow. These are:
Please find more detailed information on various steps of the workflow in the "Further reading" section to learn more about them.
Briefly, the workflow, submitted to the final_output/data/".
Important notes
-
Using genes as input: The input should be a list of gene names (each gene on a new line in a .txt file). This can be a mix of HGNC gene symbols and Ensembl gene IDs. The workflow will do the rest for you.
-
Using regions as input: The input should be a list of regions (each region on a new line with chr/startPos/endPos in tab-delimited format as a .bed file). If using a region file, the fourth column of your file can be used to provide region IDs which will be used in the workflow. If left empty, the workflow itself will generate region IDs (R###). Any overlapping regions will be merged, and the region IDs will be concatenated and used as a new region ID. If you supplied region IDs that are duplicated, the workflow will crash. Making these unique per entry will resolve the issue.
-
GRCh37 vs GRCh38: If you choose both, when you provide gene names, the workflow will use our mapping file to retrieve coordinates for both GRCh37 and GRCh38 genome builds. However, when you have regions as an input, you will need to take care of the correct locations and specify which genome build you are interested. If you are interested in both build, you will need to run the workflow twice and make sure the coordinates are correct for each build.
-
The workflow will crash if the same the final output folder already contains files with the same file name. For example, if you ran the workflow for the BRCA1 gene on day 1, and you do so unchanged on day 2 (for instance as part of a list of genes) it will crash trying to copy the final data to the output folder. This is done in order to protect your outputs from accidental overwriting - if you do not need a gene's file any more please remove it before re-running the workflow in the same location.
-
Please run the workflow on a small number of genes or regions only. The workflow has been limited ten entries. Sometimes the workflow may take up to a full day (or two) to complete, even on a small number of genes.
-
In order to save disk space, please delete the content of output folders "
cromwell-workflow-logs" and "cromwell-executions" after you have checked the run was successful. Those folders simply contain redundant data and files that can be useful for debugging when the run fails. Additionally, the content of folders "svcnvCatch_output/call_logs" and "svcnvCatch_output``/workflow_logs" can also be deleted safely after a successful run, if not needed. -
Best practices: run one query (at max ten genes or ten regions) per directory. So, that means you should copy the workflow as indicated below and rename to something relevant for each query you have.
-
If you need to re-start the workflow use
./makeClean.shfirst.
Workflow files¶
The workflow files are located in:
/gel_data_resources/workflows/BRS_tools_svcnvWorkflow/v1.2/
The workflow is contained within two files specified by the .wdl suffix. These are:
svcnvCatchCore.wdl(Main workflow script scattering over the genome build, and input regions)svcnvQueries.wdl(A sub workflow script containing the individual queries)
There is one file that specifies runtime inputs, such as the input file name, and Release Programme Version. This is:
svcnvCatchInputs.json
There is another file that specifies remaining options for the workflow, such as the output dir for overall output and output dir for log-files. This is:
svcnvCatchOptions.json
There are two files which specify which genes or regions need to be used. This is:
gene_list.txtregion_list.bed
There is a single shell script that is used to submit the workflow to the HPC. This is:
submit_script.sh(see instructions below)
There is a file detailing how Cromwell interacts with the LSF scheduler. This is:
cromwell.conf(do not edit)
Finally, there is a file containing file-paths to retrieve the gene coordinates. This is:
GRCH38_GRCH37_files.txt(do not edit)
Instructions¶
Overview¶
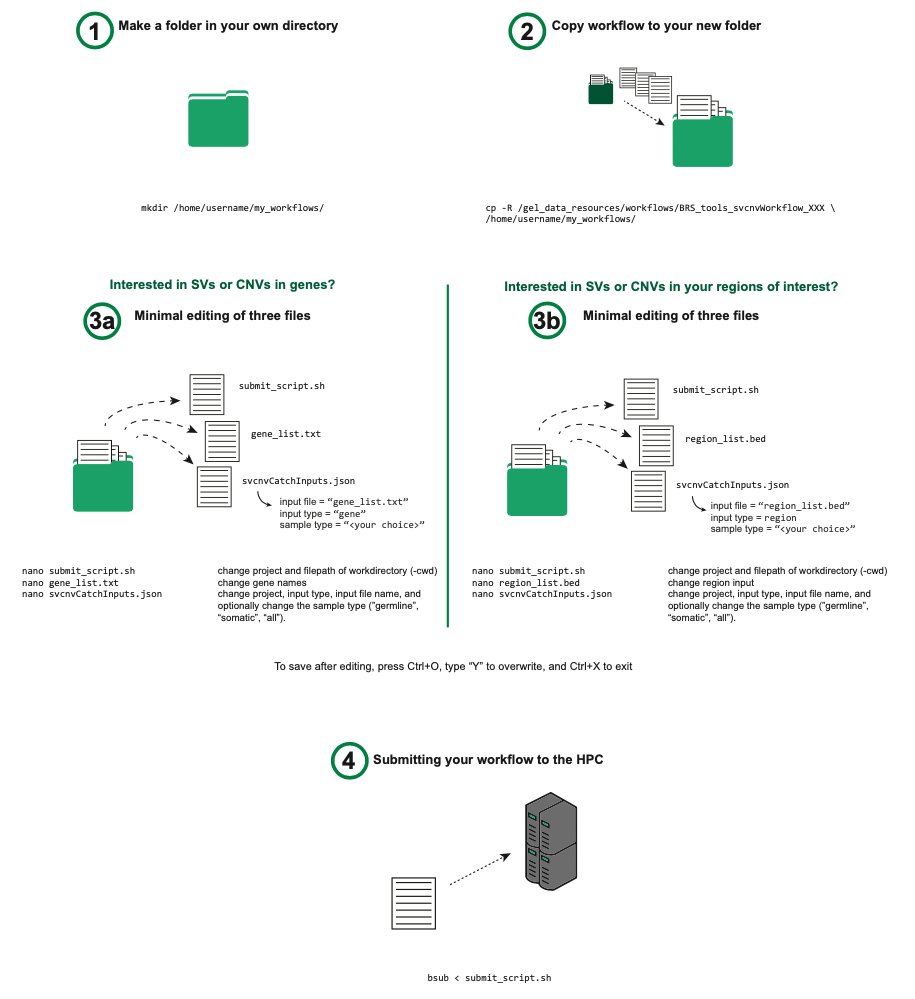
Usage (tell me what I need to change to run it)¶
What to edit to run the workflow? And how to start it?
Before you start, please check whether you have configured your R LabKey API for the HPC.
I am interested in SVs and CNVs affecting genes¶
-
Copy all the files from our
/gel_data_resources/workflows/BRS_tools_svcnvWorkflow/v1.2/folder to your own workflow folder, andcdthere. -
Edit the
gene_list.txtinput file (a .txt file containing a list of HGNC gene symbols or Ensembl gene IDs)- For example:
nano gene_list.txt- Inside gene_list.txt add the gene names of interest (here BRCA1, TNF and an Ensembl ID ENSG00000227518)
- Save the file (Ctrl+O)
-
Edit
submit_script.shto specify a valid project code. Please check your project code at LSF Project Codes. This will correspond to your respective GECIP or Discovery Forum group. -
Edit
svcnvCatchInputs.jsonto specify a valid project code, cohort type, and input type and file.- As you are interested in genes, set
svcnvCatch.input_type": "gene"and"svcnvCatch.input_file": "gene_list.txt" - Cohort type: You can specify whether you specifically want to screen germline, somatic, or both types of vcf's. Set
"svcnvCatch.sample_type": "<type>"to"germline","rd-germline","ca-germline","somatic", or"all". - Choice of genome build: You can specify if you want GRCh37, GRCh38 or both. Set
"svcnvCatch.input_genome_build": "<type>"to"both", "GRCh37", or"GRCh38". Note that all cancer samples are aligned to GRCh38. - Choice of data release: You can specify the data release. Set
"svcnvCatch.release_version": "<vesrion>"to"/main-programme/main-programme_v19_2024-10-31", or another version. - Location of reference file: You need to specify the path for your working folder where the path for the reference genomes is located. You may choose not to change it as you can read the location in /gel_data_resouces. For instance,
"svcnvCatch.GRCH37_and_GRCH38": is set to"/gel_data_resources/workflows/BRS_tools_svcnvWorkflow/v1.2/GRCH38_GRCH37_files.txt". - HPC project code: please check your project code at LSF Project Codes and update
"svcnvCatch.project"accordingly. E.g"df_yourcompanyname"OR"re_gecip_yourdomain". - Set
"svcnvCatch.dragen": "true"to query dragen aligned files. Set it to"svcnvCatch.dragen": "false", to query the default files of the 100,000 genome project.
- As you are interested in genes, set
-
OPTIONALLY, edit file
submit_script.shto specify the main working directory (the default is set to your current working directory) - note that in this case, all relative paths in config and input files need to refer to this directory:#BSUB -cwd /filepath/to/your/workflow_folder/
-
Please make sure you have read the IMPORTANT note in the "Description" section above.
-
Once you have edited all the above files accordingly, you are ready to run the script.
- In the terminal (HPC), enter the following line:
bsub < submit_script.sh
I am interested in SVs and CNVs within my region of interest¶
-
Copy all the files from our
/gel_data_resources/workflows/BRS_tools_svcnvWorkflow/v1.2/folder to your own workflow folder, andcdthere. -
Edit the
region_list.bedinput file (a tab delimited .bed file containing a your regions of interest)- For example:
nano region_list.bed- Inside region_list.bed add your regions of interest. By using the correct chromosome annotation you will retrieve results for GRCh37 (chr = ##) or GRCh38 (chr = chr##).
chr21 25800000 25900000 myRegion1_GRCh38 chr21 25910000 26000000 myRegion2_GRCh38 21 26010000 26020000 myRegion3_GRCh37 21 26030000 26040000 myRegion4_GRCh37- Save the file (Ctrl+ *
-
Edit
submit_script.shto specify a valid project code. Please check your project code at LSF Project Codes. This will correspond to your respective GECIP or Discovery Forum group. -
Edit
svcnvCatchInputs.jsonto specify a valid project code, cohort type, and input type and file.- As you are interested in genes, set
svcnvCatch.input_type": "region"and"svcnvCatch.input_file": "region_list.bed" - Cohort type: You can specify whether you specifically want to screen germline, somatic, or both types of vcf's. Set
"svcnvCatch.sample_type": "<type>"to"germline","rd-germline", "ca-germline","somatic", or"all". - Choice of genome build: You can specify if you want GRCh37 or GRCh38. Set
"svcnvCatch.input_genome_build": "<type>"to"GRCh37", or"GRCh38". Note that all cancer samples are aligned to GRCh38. Note that the workflow does not map regions between the different build and therefore for region input you cannot set input_genome_build to 'both'. - Choice of data release: You can specify the data release. Set
"svcnvCatch.release_version": "<vesrion>"to"/main-programme/main-programme_v19_2024-10-31", or another version. - Location of reference file: You need to specify the path for your working folder where the path for the reference genomes is located. You may choose not to change it as you can read the location in /gel_data_resouces. For instance,
"svcnvCatch.GRCH37_and_GRCH38": is set to"/gel_data_resources/workflows/BRS_tools_svcnvWorkflow/v1.2/GRCH38_GRCH37_files.txt". - HPC project code: please check your project code at LSF Project Codes and update
"svcnvCatch.project"accordingly. E.g"df_yourcompanyname"OR"re_gecip_yourdomain".
- As you are interested in genes, set
-
OPTIONALLY, edit file
submit_script.shto specify the main working directory (the default is set to your current working directory) - note that in this case, all relative paths in config and input files need to refer to this directory:#BSUB -cwd /filepath/to/your/workflow_folder/
-
Please make sure you have read the IMPORTANT note in the "Description" section above.
-
Once you have edited all the above files accordingly, you are ready to run the script.
- In the terminal (HPC), enter the following line:
bsub < submit_script.sh
SV and CNV queries details
All queries filter for variants with filter PASS.
Cancer queries include only tumour samples, excluding the matched germline samples.
Samples with more than one alignment had only the most recent one returned.
-
SV queries:
all SV types are retrieved: duplications (DUP), deletions (DEL), inversions (INV), insertions (INS) and translocations (BND)
output format:germline: [%SAMPLE] %CHROM %POS %ID %REF %ALT %QUAL %FILTER %INFO/SVTYPE %INFO/SVLEN %INFO/END [%GT] [%GQ] [%PR] [%SR]
somatic: [%SAMPLE] %CHROM %POS %ID %REF %ALT %QUAL %FILTER %INFO/SVTYPE %INFO/SVLEN %INFO/END [%PR] [%SR] %CSQT %CSQR %cosmic %clinvar
-
CNV queries:
CNVs with copy number different than 2, and CN = 2 where loss of heterozygosity (LOH) have been observed are retrieved.
output format:all: [%SAMPLE] %CHROM %POS %ID %REF %ALT %INFO/END %INFO/SVTYPE [%RC] [%BC] [%CN]
Inputs and outputs¶
| Type | Type | File or Folder | Description | Requires editing |
|---|---|---|---|---|
| Input | File | gene_list.txt | .txt file containing a list of HGNC gene symbols or Ensembl gene IDs (new line separation). | yes (for genes as input) |
| Input | File | region_list.bed | .bed file containing chr/start/end positions for the region of interest | yes (for regions as input) |
| Input | File | svcnvCatchCore.wdl | Main workflow script. | no |
| Input | File | svcnvQueries.wdl | A sub workflow script, scattering over genome build. | no |
| Input | File | svcnvCatchInputs.json | Specifies runtime inputs such as the input file name and type, cohort type, genome build and Release Programme Version. | yes (see instructions) |
| Input | File | svcnvCatchOptions.json | Specifies output dir for overall output and output dir for log-files. | no (optional) |
| Input | File | cromwell.conf | Configuration file on how Cromwell interacts with the LSF scheduler. | no |
| Input | File | GRCH38_GRCH37_files.txt | Contains the locations of files with gene identifiers and their respective regions. | no |
| Input | File | submit_script.sh | The final submission script to run the workflow. | yes (for project code and optionally working directory) |
| Output | File | svcnvCatch_output/data/adjustedInput~{genome_version}~{timestamp}.tsv | The file that is created based on your input. This is used further downstream in the workflow. Any duplicate regions are filtered out and merged (see below). | NA |
| Output | File | svcnvCatch_output/data/CNVmergedFailed~{queryID}___~{genome_version}~{cohort_type}~{timestamp}_.tsv | Contains filepaths of vcf's that could not be accessed during the workflow run. | NA |
| Output | File | svcnvCatch_output/data/CNVmergedResult~{queryID}___~{genome_version}~{cohort_type}~{timestamp}_.tsv | Contains all the positions of CNVs detected for the input region or gene (=queryID). | NA |
| Output | File | svcnvCatch_output/data/SVmergedFailed~{queryID}___~{genome_version}~{cohort_type}~{timestamp}_.tsv | Contains filepaths of vcf's that could not be accessed during the workflow run. | NA |
| Output | File | svcnvCatch_output/data/SVmergedResult~{queryID}___~{genome_version}~{cohort_type}~{timestamp}_.tsv | Contains all the positions of SVs detected for the input region or gene (=queryID). | NA |
| Output | File | svcnvCatch_output/data/coordinates~{genome_version}_.bed | Coordinates file generated and used by the workflow. | NA |
| Output | File | svcnvCatch_output/data/logfile~{timestamp}.log_ | A log file that is generated within the workflow containing all the versions of the software and settings that were used. | NA |
| Output | Folder | svcnvCatch_output/data/ | Will contain the data output from the workflow when it successfully completes. | NA |
| Output | Folder | svcnvCatch_output/call_logs/ | Contains all the log files that are generated during the query. | NA |
| Output | Folder | svcnvCatch_output/workflow_logs/ | Will contain the running workflow log. | NA |
| Output | Folder | cromwell-workflow-logs/ | Will contain the overall workflow log. | NA |
| Output | Folder | cromwell-executions/ | All the temporary files for each run are stored within the genetic_report folder. Once the jobs have been completed, they will be copied over to the final output folder specified by the user. This will also contain the cromwell database for callcaching. In case an identical query is being done, the workflow will check this database to see if it has been done before, and simply extract the data again without running the full script again. |
NA |
Output breakdown¶
Adjusted Input file (adjustedInput_~{genome_version}_~{timestamp}.tsv):
If your input were a set of regions, this file will only contain the first four columns, where the fourth column are region identifiers.
| Column | Description |
|---|---|
| chr | chromosome |
| start | start position of gene region |
| end | end position of gene region |
| gene | HGNC gene symbol |
| ensemblID | Ensembl gene ID |
| internalID | identifier generated internally which will be used as a prefix for output files: chr_gene_ensemblID |
| posTag | position tag (chromosome_start_end) aiding the identifier deduplication step |
| originalInput | specifies what has been used as an input by the user, either "gene" or "ensembl", referring to either gene symbol or Ensembl ID |
| entryDuplicate | NA, or containing a comma-separated string of internalIDs. The internalIDs listed here are considered the same entry due to their identical genomic position. |
Coordinates file
coordinates_~{genome_version}.bed:
Coordinates bed file used by the workflow.
Unrecognised input file unrecognised_input_~{genome_version}_~{timestamp}.tsv:
Similar to the input file specifying the entries which were not detected in the flat file containing all gene and Ensembl IDs. This file will not be generated if all inputs were valid, nor if you selected to use regions as an input.
SV Merged Result SVmergedResult__~{queryID}_~{genome_version}_~{cohort_type}_~{timestamp}_.tsv:
| Column | Description |
|---|---|
| platekey | platekey ID of participant |
| chr | Chromosome |
| pos | Start position of the variant |
| ID | Variant ID |
| REF | Reference allele |
| ALT | Alternate allele |
| Qual | quality |
| Filter | PASS |
| INFO/SVTYPE | Type of structural variant: duplication (DUP), deletion (DEL), inversion (INV), insertion (INS) or translocation (BND) |
| INFO/SVLEN | Variant size |
| INFO/END | End/Continuation position of the variant |
| GT | [germline] Genotype. 0/1 = heterozygote, 1/1 = homozygote |
| GQ | [germline] Genotype quality |
| PR | Spanning paired-read support for the ref and alt alleles in the order listed |
| SR | Split reads for the ref and alt alleles in the order listed, for reads where P(allele |
| CSQT | [somatic] Consequence type as predicted by IAE. Format: GenotypeIndex |
| CSQR | [somatic] Predicted regulatory consequence type. Format: GenotypeIndex |
| cosmic | [somatic] The numeric identifier for the variant in the Catalogue of Somatic Mutations in Cancer (COSMIC) database. Format: GenotypeIndex |
| clinvar | [somatic] Clinical significance. Format: GenotypeIndex |
CNV Merged Result CNVmergedResult_~{queryID}_~{genome_version}_~{cohort_type}_~{timestamp}.tsv:
| Column | Description |
|---|---|
| platekey | platekey ID of participant |
| chr | Chromosome |
| pos | Start position of the variant |
| ID | Variant ID |
| REF | Reference allele |
| ALT | Alternate allele |
| INFO/END | End position of the variant |
| INFO/SVTYPE | CNV for variants with copy number != 2. LOH for variants with copy number = 2, and loss of heterozygosity |
| RC | Average counts per bin |
| BC | Bin count |
| CN | Copy number |
CNV Failed Result (
CNVmergedFailed_~{queryID}_~{genome_version}_~{cohort_type}_~{timestamp}.tsv:
This file will contain filepaths from vcf's that could not be accessed. If everything was accessed, it will contain a message stating this.
SV Failed Result SVmergedFailed_~{queryID}_~{genome_version}_~{cohort_type}_~{timestamp}_.tsv:
This file will contain filepaths from vcf's that could not be accessed. If everything was accessed, it will contain a message stating this.
Plots¶
Version v1.1.1 or later outputs plots for each gene/region, somatic/germline and genome build. For each of these combinations a .pdf file is generated for SV and CNV separately. On the plots, the right y-axis summarises vcf information. N is the number of participants with SV/CNV that start and end at the same position. The remaining of the fields give the median across all samples that have that variant. Grey variants do not overlap CDSs while the one in colours do.