Rare disease tiering¶
The Genomics England Rare Disease Tiering Process identifies possibly pathogenic variants in affected probands.
The goal of tiering is to aid NHS GMC evaluation of Rare Disease primary finding results by annotating variants that are plausibly pathogenic based on their segregation in the family, frequency in control populations, effect on protein coding, mode of inheritance, and whether they are in a gene in the virtual gene panel(s) applied to the family. The process is summarised in the figure below.
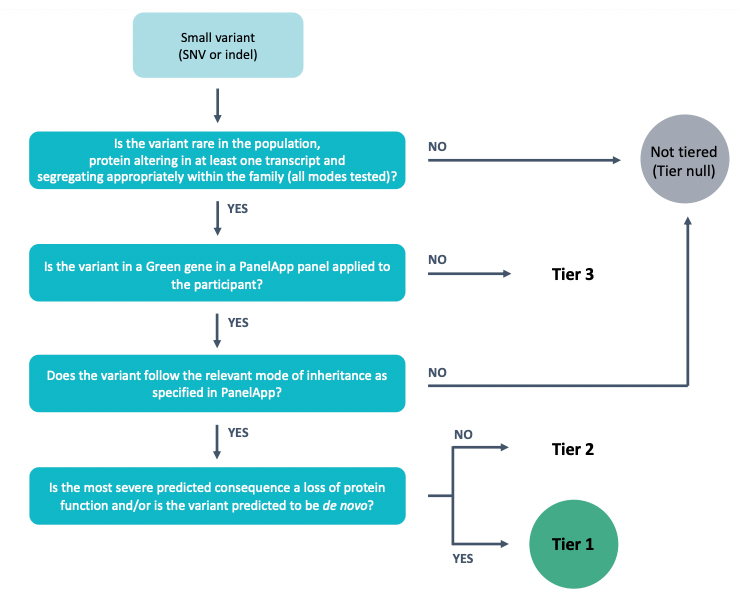
During the Tiering process, variants (which have been previously called, normalised, and annotated by the Rare Disease Interpretation Pipeline) pass through multiple filters (allele frequency, consequence type, segregation, quality etc.) in order to classify those that are potentially relevant/causal for a specific case and disease.
A similar process is also used to classify variants in the Cancer Programme. The implementation of Tiering for the two programmes are very different. This documentation is focused only on the Tiering process for Rare Disease.
Variants of potential relevance to the patient’s clinical presentation will be automatically categorised into three Tiers:
-
TIER 1: Should be clinically assessed by NHS GMCs. Includes, high impact variants (e.g. likely loss-of function) and de novo moderate impact variants (e.g. missense) within a curated list of genes available through PanelApp with sufficient evidence associating them with the patient’s phenotype(s). In the future it is anticipated Tier 1 will contain known pathogenic variants once a high confidence curated list has been generated.
-
TIER 2: Should be clinically assessed by NHS GMCs. Includes moderate impact variants (e.g. missense) within a curated list of genes available through PanelApp with sufficient evidence associating them with the patient’s phenotype(s).
-
TIER 3: It is not expected that NHS GMCs will review all of the variants in Tier 3. Plausible candidate variants identified in genes OUTSIDE of known disease gene panel(s), caution should be used during clinical assessment and interpretation.
Includes high and moderate impact variants outside of the curated list of genes that are associated with the patient’s phenotype(s). Although most tier 3 variants will NOT be pathogenic, sometimes the causal variant will lie within tier 3. This could occur because there is insufficient evidence to support the inclusion of the gene within the relevant panel(s) at the time of analysis, or because the relevant panel was not applied.
You can learn more about the Tiering algorithm in the Rare Disease Results Guide and FAQs below.
Location¶
Tiering data¶
Tiered variants for interpreted rare disease families can be found within the LabKey table, tiering_data. Within this table, you can filter variants by: Tier (Tier 1, Tier 2, Tier 3), chromosomal position, gene name, variant consequence type, genotype, and phenotype, as well as other variables. You can also browse the raw Tiering data in the Interpretation Request file.
Allele frequency of tiered variants¶
The LabKey table, tiered_variants_frequency, displays the allele frequency of all Tiered variants in various cohorts.
Gene panels applied¶
The LabKey table, panels_applied, displays the gene panels applied to the family during the Tiering process and the panel version. You can browse all panels and versions in PanelApp.
FAQs¶
Here are some commonly asked questions about the tiering data. If you have more questions, please raise a ticket at the Genomics England Service Desk.
How do clinicians select the panels for a participant?
Clinicians identify the relevant panels using the National genomic test directory.
How do we decide what variant falls under denovo_segregation within the Tiering Data?
Variants are only marked as denovo when both parental genomes were available in the family-wide variant calling and where the respective variants were not present in either of these genomes and only observed in the off-spring.
In case you wish to verify specific denovo assignments listed in the tiering_data table, you are now able to check this through the use of the rare_disease_interpreted table. The platypus_vcf_path column will provide you with the file path for the family-wide vcf (Platypus pipeline) that was used for the interpretation, and therefore the tiering_data table.
In case a relative has withdrawn their consent, the data may not be fully available. Please reach out to the Genomics England Service Desk to verify whether a participant has been withdrawn.
During the Genomics England Tiering process, variants are filtered for rare variants prior to being permitted to the tiering system. What frequency thresholds are used to determine the "Rare Variant" status?
During the Tiering process, variants (which have been previously called, normalised and annotated by the Rare Disease Interpretation Pipeline) pass through multiple filters (allele frequency, consequence type, segregation, quality etc.) in order to classify those that are potentially relevant/causal for a specific case and disease. The allele frequency mentioned here refers to the frequency in control populations. These allele frequencies and populations can be found in the Rare Disease Results guide under section 5.3.4 Tiering Algorithm Criteria, criterion 2 (page 14).
I observed some duplicate variants within the same participant? What does this mean?
Generally speaking, there should be just one row for a given variant. However, some rows or variants may appear twice in the table due to data wrangling the tables from JSON (in our database) to flat file (for LabKey and general usage). However, some variants can be in different impact categories for instance or modes of inheritance which in turn causes the partial "duplication" of the rows.
If a variant is categorised as CompoundHeterozygous segregation (and heterozygous), how do we know which the "second" variant of the compound heterozygous pair is?
Compound heterozygotes are determined within the space of the protein coding region of a gene (and potentially around splicing sites). When this occurs you should see at least one other variant within that gene also being listed as CompoundHeterozygous. It is possible that you will see >2 compound heterozygotes in a gene which can then be considered as one group. Unfortunately, it is unknown whether how each of the alleles affect each other and the disease type so we cannot provide more detail beyond the groups of compound heterozygotes.
Why are genes or variants listed in multiple tiers? And how do I know which one is correct?
Similar to the question above, variants can occur as multiple rows for a single participant. One of the reasons for this can be that the gene is part of multiple tiers, and each variant by tier is displayed on a different row. Therefore, variants can be listed with multiple tiers, and effectively both are correct. For the tiering process, each phenotype will have various gene panels applied to them. Depending on the gene panels that are being applied, some genes may be present and some may not.
For instance, your gene of interest was listed as a "Green" gene in one of the gene panels, and therefore passes various downstream thresholds resulting in its classification as a potential protein truncation/de novo mutation. However, the same gene may not be considered as a "Green" gene in another specific panel, and thus ended up as a Tier 3 gene. To find out which gene panels are applied for a participant and phenotype, you can look at the panels_applied table in LabKey.
This does raise the question as to which Tier relates to which panel as in some cases a single phenotype can have up to nine panels applied to it. Unfortunately we do not provide that information in the tiering_data table, but this may change in the future though this is not formally on the horizon. Because of this gap, we either use both tiers or select the most impactful tier depending on the analysis that we perform as ultimately the specific gene panels do relate to the phenotype of the individual.
What parameters have been applied for the family-based variant calling which is being used by the tiering process?
Our tiering process uses VCF' that have been generated by the platypus pipeline performing family-based variant calling. While we are unable to provide these run options publicly available, you are able to retrieve them within the Research Environment, or by raising a Genomics England Service Desk request. While the run options between the different cohorts and family are essentially the same (beyond different reference genomes), the approach below is aimed at retrieving the options for a single family.
-
Open the
rare_disease_interpretedtable (Data Release 12, or higher). -
Filter for the family of interest
-
Retrieve the file path from the column "Platypus Vcf Path", which should follow the following structure:
/gel_data_resources/main_programme/platypus_interpreted_vcf/<genome_build>/<interpretation_id>/<proband_platekey>.duprem.atomic.left.split.vcf.gz -
Open the terminal application and proceed using the code below for either the RE or via the HPC
For more general information on the platypus pipeline and what individual parameters mean, please have a look at the following external page: https://www.rdm.ox.ac.uk/research/lunter-group/lunter-group/platypus-documentation